Founder | Pharmaceutical Professional
✓ Reviewed by: Pankaj Sharma - Quality Control Specialist
Reviewed for Quality Control accuracy, laboratory practices, and technical relevance
📅 Last Updated: September 23, 2024
HPLC instruments are widely used in pharmaceutical, chemical, and other industries to separate, distinguish, or quantify each component in a mixture. As a result, HPLC-related jobs are in high demand, and interviews for these positions can be competitive. To increase your chances of success, it’s important to prepare thoroughly for HPLC interviews. In this article, we will discuss and provide you with a complete overview of HPLC interview questions and answers to help you crack your next job interview.
What is HPLC?
HPLC is a high-performance liquid chromatography. This technique is used to separate, identify, and quantify a mixture of components. Suppose I have a mixture of components present, and different types of components are present. So to separate and identify a component from a mixture of different components, for example, if I have separated the protein component, I have identified that, it is just a protein molecule and quantified that, it is present in this much amount in the sample. So the technique used for this is high-performance liquid chromatography.
This is a type of liquid chromatography. In liquid chromatography, the mobile phase used is liquid, That’s why we call it liquid chromatography. While high performance tells us that the resolution power of HPLC is very good. Resolution power means the ability to distinguish between two components. If there are different components, it can distinguish very well, That’s why this technique is used the most in labs.
The development of column chromatography from low-pressure compatible glass columns to high-pressure compatible metal columns occurred in the 1960s. So, this was the short intro, now let’s get into the HPLC Interview Questions and Answers series.
Mostly asked HPLC Interview Questions and Answers
Q. Define HPLC
Ans: HPLC is a high-performance liquid chromatography technique used to identify, quantify, and separate the compounds from a mixture.
Q. What is the Principle of HPLC?
Ans: It is a technique used to separate the mixture of components based on the adsorption, partition, ion exchange, and size exclusion principle.
Q. Phase use in HPLC
Ans: Mobile phase and stationary phase
Q. What is used to transfer the MP from one module to another?
Ans: The mobile phase is transferred through stainless steel capillaries.
Q. Tell me the internal diameter of the capillaries.
Ans: The internal diameter between the pump and injector is around 0.5 to 1 mm and 0.2 mm in the case of capillaries after the outlet of the injector.
Q. Why is HPLC also known as high-pressure liquid chromatography and liquid chromatography?
Ans: In high-performance liquid chromatography high pressure is applied through a pump to move the mobile phase through the column.
high-performance liquid chromatography is also known as Liquid chromatography because the mobile phase used is liquid.
Related Post: Technical Pharmaceutical Interview Questions/Answers
Q. Chromatographic condition for use in HPLC analysis?
Ans: Chromatography condition means method, it consists of the following parameters.
- Mobile phase composition
- Types of column
- Wavelength
- Flow rate
- Injection volume
- Run time
Q. What is chromatogram in HPLC?
Ans: The CHromatogram is a plot of detector signal output versus time or elution volume.
Q. What is the retention time in high-performance liquid chromatography analysis?
Ans: The time taken by the analyte peak to reach the detector is called retention time.
Q. What are the mobile phase and stationary phase in high-performance liquid chromatography?
Ans: The mobile phase is the liquid that moves the solute through the column. and the stationary phase is the packaging material of the column which is the immobile phase involved in the chromatographic process.
Q. What is the separation mechanism in HPLC?
Ans: Compounds are separated because the molecules move at different rates in the column due to different interactions between the stationary phase and sample, the molecules move at a different rate, therefore separation can be done.
Q. What is the mode of high-performance liquid chromatography?
Ans:
- Normal phase chromatography
- Reverse phase chromatography
- Ion chromatography
- Size exclusion chromatography
- Affinity chromatography
Q. High-performance liquid chromatography types On the basis of the elution technique
Ans: Isocratic separation
Gradient separation
Q. What is the USP general chapter number for the chromatography?
Ans: In the United States Pharmacopeia (USP) refer to general chapter no. <621> for chromatography.
Q. What are the reverse phase and normal phase modes?
Ans: In Reverse phase mode: Stationary phases, non-polar mobile phase, and polar
In Normal phase mode: Stationary phase, polar mobile phase, and non-polar.
Q. What is a frit in HPLC?
Ans: Frit is the inert physical device, typically installed before pre-columns, that provides a course filtration of the HPLC eluent and prevents containment particles from reaching the high-performance liquid chromatography System.
Q. What is the cause of baseline drift or noise?
Ans: Baseline drift often appears when there is a temperature fluctuation especially with refractive index and conductivity detectors or with UV detectors at high sensitivity.H
A mobile phase mixing chamber not working properly will account for an uneven mobile phase causing baseline drift as well. contamination or air buildup in the detector cell could also be responsible for baseline drift/noise.
Related Topic: Quality Assurance Interview Questions Pharma
Q. What is the main component of HPLC?
Ans:
- Solvent delivery pump
- Injector
- Column oven
- Detector
Q. Based on the elution technique, high-performance liquid chromatography is how many types?
Ans: On the basis of the elution technique high-performance liquid chromatography is of two types:
- Isocratic system
- Gradient system
Q. What do you mean by gradient system?
Ans: A gradient system is when two or more solvents are used for elution for analysis termed as gradient system.
Q. What is the pre-analysis check in high-performance liquid chromatography?
Ans:
- Pass the mobile phase through a filter and allow the mobile phase to come to room temperature.
- Purse all the flow lines to remove air bubbles.
- If the buffer is used as a mobile phase, wash the back of the plunger seal.
- Check for leaks
- Check the pump pressure and perform a baseline check.
Q. What are the possible reasons, why there is no peak eluted in your analysis?
Ans:
- The sample is too diluted
- The injection volume is too small
- The detector lamp is off
- the pump is not pumping
- Incorrect mobile phase and sample
- Column retains all compounds
Q. What are the detectors used in HPLC?
Ans:
- PDA- Photodiode array detector
- RF- Fluorescent Detector
- ECD- Electrochemical detector
- UV-VIS- Ultraviolet/ visible detector
- COD- Conductivity detector
- RID- Refective index detector
Q. What is the advantage of PDA detectors in high-performance liquid chromatography?
Ans:
- A PDA Detector could perform analysis on one sample at different wavelength levels without having to repeat the process.
- PDA detectors are useful for compound identification and checking peak purity.
- Relatively robust to temperature fluctuations and flow rate.
Q. Why Mobile phase degassing is important in HPLC analysis?
Ans: Several gases are insoluble in organic solvents, when high pressure is pumped, the formation of gas bubbles increases which interferes with the separation process, steady baseline, and shape of the peak.
Q. Why HPLC is a very useful technique from other chromatographic techniques?
Ans: This technique is useful because of the following reasons:
- High resolution
- High sensitivity
- Good repeatability
- Small sample size
- Moderate analysis condition
- No need to vaporize samples like GC
Q. What is the resolution factor in HPLC and how to calculate?
Ans: The resolution of elution is a quantitative measure of how well two elution peaks can be differentiated in chromatographic separation. it is measured by the difference in retention times between the two peaks, divided by the average width of the elution peak.
Q. Why do we use a gradient system in HPLC analysis?
Ans:
- Gradient systems are used in HPLC analysis because in gradient system mode it is easy to separate complex samples.
- This method gives increased sensitivity
- It gives high-resolution
Q. What is the main possible reason for the spike baseline and what are the solutions?
Ans: Bubble in the flow cell
Q. What are the remedies/solutions for spike baseline?
Ans:
- Apply back pressure to the flow cell
- De-Gas mobile phase
- Clean flow cell with IPA
Q. What is the ODS and BDS column?
Ans: ODS and BDS are two columns used for reverse-phase chromatography. the key difference between the ODS and BDS columns is that the ODS column contains Free- OH functional groups, whereas the BDS column contains deactivated -OH groups. Furthermore, BDS columns are made to reduce peak tailing while ODS columns have strong peak tailing.
Q. Differences between Empower 2 and 3 in HPLC
Ans: There are a few variations between Empower 2 and 3, the biggest one being that with Empower 3, you may combine multiple injections, rearrange the raw data, and combine the number of injections. For instance, a sample line with five injections can be divided into five separate lines. Additionally, Empower 3 permits you to estimate signal-to-noise using a blank injection in your sample set.
Empower 3 has additional system rules, such as requiring ID numbers in view filters for clarity. In Empower 3, you may watch a live injection being reviewed by selecting Plot< Update Display, too. In Empower 3, the Global Project View feature enables the review and comparison of project data such as channels and results across all projects rather than just one.
The primary functionalities, including the development and use of custom fields, data collection, and processing, are nearly identical to those of Empower 2.
Q. What is the parameter of HPLC Calibration?
Ans:
- Flow rate accuracy (Pump module)
- Injector accuracy
- Carryover
- System precision
- Wavelength accuracy
- Injector linearity
- Detector linearity
- Gradient performance check
- Column oven temperature accuracy
Q. Which standard is used for HPLC Calibration?
Ans: The caffeine standard is used for calibration.
Q. Why Caffeine is used in HPLC?
Ans:
- Caffeine is easily available in the market.
- In general, we all use caffeine as a standard for the calibration, especially for (Calibration of Wave Length Accuracy) since it will scan the entire UV range, giving a response with multiple wavelengths, including the first maximum at 205 nm, second maxima at 273 nm, and minima at 245 nm.
- It is very stable, we can use a long time for analysis i.e. it is not degradable
Fewer HPLC interview questions and answers for freshers are given below:
Q. What is SST?
Ans: A chromatographic system’s column efficiency, resolution, and repeatability are checked using the SST (also known as the System Suitability Test) analytical process to make sure it is suitable for a given analysis. Using this, it is quite simple to verify the following parameters:
- Retention time
- Pressure
- Tailing factor
- Plate number
- Column efficiency
- Resolution
- Signal-to-noise ratio
- Repeatability
- Relative Standard Deviation (RSD)
All parameters are described below:
Q. What is RRT in HPLC?
Ans: RRT means Relative retention time. it is the comparison of the retention time of one component to another.
Q. What is Relative standard deviation (RSD)?
Ans: RSD is a measure of the deviation of a set of numbers disseminated around the means.
Q. % RSD (Relative standard Deviation)/ Repeatability
Ans: % Relative standard deviation is defined as standard deviation expressed as the percentage of the mean. in HPLC 5 or 6 replicate injections of standard preparation are injected and the %RSD is calculated by following the formula.
%RSD= SD/X*100
Whereas, SD= Standard deviation and X= is the mean (requirement 2% or less)
Q. What is RRF?
Ans: RRF means relative response factor, it is a ratio of response of an equal amount of the impurity and the drug substance.
Q. What is Retention time?
Ans: The retention time (tR) is measured from sample injection (the start of the chromatogram) to the apex (high point) of the peak. it is measured in minutes.
 3")
Q. What is adjusted retention time?
Ans: (t’R) = tR – tM
Where:s
tR = retention time of the given component
tM = retention time of sample solvent, Measured in minutes
Q. What is Peak Width?
Ans: Baseline width is obtained manually by drawing the tangents to the inflection points on the front and back slopes of the peaks and then measuring the distance between the points where those tangents intersect the baseline.
 4")
Q. What is Resolution?
Ans: Resolution is defined as the ratio of the center-to-center separation between two adjacent peaks to the average baseline width of those peaks.
 5")
Q. Measurement of the degree of separation and column efficiency:
Ans: R = [tR(B) – tR(A)] / [1/2(WB(A) + WB(B))]
By definition, Rs > 1.5 is considered a “baseline” resolution.
Resolution Example:
 6")
 7")
 8")
 9")
 10")
 11")
Q. What is the Asymmetry factor?
Ans: The Asymmetry Factor (As) is commonly used in non-pharmaceutical applications. It is calculated by dividing the center-to-back distance by the center-to-front distance, as shown on the right, with both measurements made at 10% of the maximum peak height.
Q. What is the Tailing Factor?
Ans: The Tailing Factor (TF) is specified by the United States Pharmacopoeia (USP)
Calculated by dividing the front-to-back distance by twice the center-to-front distance, with both measurements made at 5% of the maximum peak height.
 12")
- A perfectly symmetrical peak has an As or TF of 1.0.
- As or TF reaching a value of 1.5 and below are generally said to be within the “acceptable” limit.
- As or TF values in excess of 2.0 are generally considered unacceptable.
- It is possible (but not common) to have As or TF values less than 1.0 (i.e., the shallow slope is on the leading edge of the peak rather than the trailing edge). As or TFs below the value of 1.0 are referred to as “fronting”.
Q. What is Peak Area?
Ans: The area under the peak is directly proportional to the mass of the analyte injected. One Type of peak response suitable for a wide range of applications
Q. What is Peak Height?
Ans: Peak Height is indirectly based on peak area.
If the width and asymmetry of a peak are constant from run to run, then the height of that peak will be directly proportional to its area.
Peak height is more convenient for manual measurement and may provide more reproducibility for very small peaks or very noisy baselines.
 13")
Q. Column efficiency/ How Measurement of the column efficiency:
Ans: The chromatographic column contains a large no. of separate layers called theoretical plates.
N the no. of the theoretical plate is used to determine the performance and effectiveness of the column and it’s calculated using the following formulas:
Gaussian peak/electronic integrator formula
Number of theoretical plates = N
 14")
t- retention time
w- peak width
Q. HETP = H
Ans: H = column length / N
Q. Capacity factor, k’
Ans: The capacity factor also called “Capacity Ratio” is symbolized by k’. it is a measure of the retention of the peak that is independent of column geometry or mobile phase flow rate.
The optimum value for k’ is 2-6 minutes. The larger the k’, the better the resolution
Selectivity, a (Indicates quality of separation)
 15")
a = 1, no separation of A and B.
a = 2, good quality separation.
Q. Signal-to-noise ratio (S/N ratio)
Ans: The signal-to-noise ratio in liquid chromatography separation is measured between two lines bracketing the baseline and the signal from the middle of the baseline to the top of the peak.
The formula for signal-to-noise ratio: S/N= 2H/h
Q. Reference standard check (Similarity factor)
Ans: Two standard solutions are prepared (A&B). check the accuracy of the solution preparation. similarity factor should be 0.98 to 1.02.
The formula for Reference standard check (Similarity factor):
 16")
Related Job: Dissolution Interview Questions with Expert Answers from Pharma Professionals
Common HPLC Interview Questions and Answers
Q. Why do we need all the complicated high-tech equipment like HPLC? or Advantages of HPLC
Ans: Advantages are the following:
Speed: A “Classical” LC analysis can take between 2 to 12 hours to complete. It is possible to carry out a comparable analysis within a period of 2 to 12 minutes by HPLC.
Reproducibility: For each analysis, a classical column must be freshly packed, increasing the possibility of error. Numerous samples can be run through a single HPLC column at once.
Quantitation: During the separation process, HPLC includes on-line detectors that give quantitative information during the separation process.
Sensitivity: Classical LC made use of relatively large columns and considerable amounts of mobile phase solvents. The resulting dilution reduced the method’s sensitivity. Miniaturized columns are used in HPLC to minimize sample dilution.
Column: Band broadening is minimized by reducing the average particle size of the column packing. A similar reduction in column size reduces solvent consumption and sample dilution.
Pump: Because only gravity can not propel liquid through a very fine particle column at a reasonable velocity, a constant flow pump must be employed.
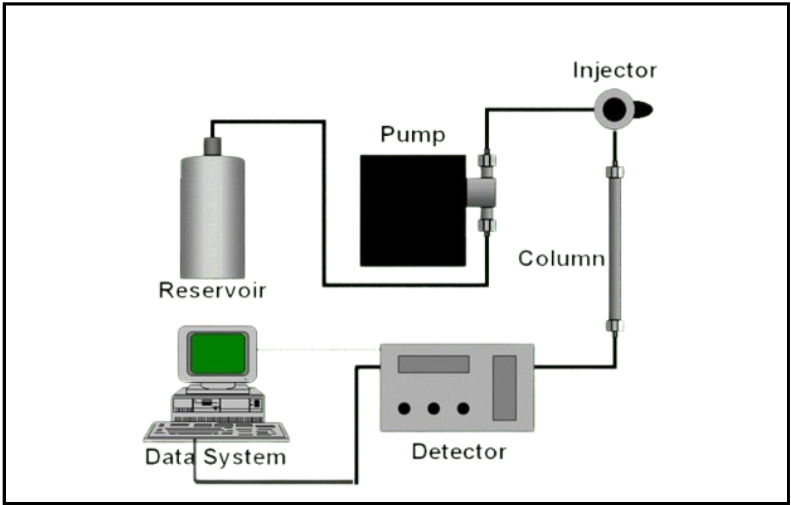
Reservoir: A reservoir must be present to ensure a regular supply of mobile phase (MP).
Detector: It is pointless to stand and collect and then separately analyze fractions as it involves a lot of work. Hence, some kind of flow-through detector is relied upon to detect the presence of sample compounds in the column effluent.
Data System: A data system is ultimately needed to compile and analyze the findings.
This used to be frequently a straightforward strip-chart recorder or a standalone integrator. Today, a dedicated PC-type computer is more likely to be the data system.
Related Topic: Pharma Packing Questions for the Interview
Q. End result in High-performance liquid chromatography?
Ans: With six basic modules
- Reservoir
- Pump
- Injector
- Column
- Detector, and Data system.
Troubleshooting problems HPLC interview questions and answers:
Q. What do you mean by troubleshooting?
Ans: Troubleshooting is a form of repairing or thinking about repairing, troubleshooting is often done to repair failed procedures or processes on instruments. This is the process of finding a problem and trying to locate its cause, in order to fix the problem and bring the operation of the process, product, or system back on track.
Q. What are the six steps of troubleshooting problems?
Ans:
- Identify the problem
- Established a theory of probable cause
- Test the problem cause theory to determine the actual cause.
- Established an action plan and executed the plan.
- Verify full system functionality.
- Document the process at every stage.
Q. What causes RSD failure in HPLC?
Ans:
- The most common causes of HPLC failure:
- Column temperature fluctuations.
- No/low pressure are solvent inlet lines not being immersed in the solvent.
- No solvent in the reservoir and leaks.
- Air trapped in the pump.
- Sample solvent incompatible with the mobile phase.
- The mobile phase is not properly degassed.
- Column problem.
Q. What happens if your sample solvent is stronger than your mobile phase?
Ans: A more potent solvent is not advised because of the tendency of its injection to cause peak dystopia which includes broadening of the peak with poor sensitivity and short retention times. this happened because some analytes will tend to travel too quickly through columns, instead of eluting in a symmetrical band.
If you absolutely must do this, keep the volume as small as possible and make sure the solvent is miscible.
Q. What to do when the backpressure increases?
Ans:
- This could be a guard or an analytical column issue. to exactly pin point where the problem occurs, we suggest you get rid of the guard column (if you are using one) and replace the old cartridge with a new one. if the original pressure is back to normal, congratulations, you have solved the problem.
- Disconnect the analytical column from the system and backflush it if the pressure remains high (do not connect the column to the detector while doing so.), and run small column volumes of your MP through the column.
- If the problem still persists you may have some strongly retained contaminants in the column coming from previous injections. run the appropriate restoration procedure, as suggested by the column manufacturer, and retest the column.
- Change the inlet frit or replace the column if the initial pressure is not restored.
Q. What are the causes of baseline drift/ noise?
Ans:
- Baseline drift often appears when there are temperature fluctuations (small variations can cause important baseline drift) especially with refractive index and conductivity detectors or with UV detectors at high sensitivity.
- A mobile phase mixing chamber not working properly will account for uneven mobile phases causing baseline drift as well.
- Contaminants or air build-up in the detector cell could also be responsible for baseline drift/noise.
Q. Tell me the Five probable causes of variable retention times.? (RT shift)
Ans:
- Due to leaks or changes in mobile phase composition (a small change in the mobile phase can lead to large changes in retention time.
- Air trapped in pump: Trapped air in pump heads can also increase or decrease retention time changes.
- Fluctuations in Column Temp: Column temperature fluctuations, especially in ion exchange chromatography, can also lead to retention time changes.
- Contaminated mobile phase: A contaminated mobile phase will also cause the column retention time to shift.
- Retention time: Retention time usually decreases when the mass of the solute exceeds the column capacity and has a tendency to increase with volume overload.
More Helpful, HPLC interview questions and answers in Pharmaceuticals are given below:
Q. What to do when there are no peaks or small peaks?
Ans:
- The first thing that should be checked is the on status of the detector lamp.
- Check the wire between the detector and the integrator. A loose or broken wire could also be the source of the problem.
- Make sure the pump is on, the flow is towards the column and not going to your waste.
- Check the sample, and be sure it does not deteriorate. check for bubbles in the vials.
- We make sure no air bubbles in sampler vials and automatic sampler vials have sufficient liquid. evaluate system performance with fresh standards to confirm the sample as a source of problems.
Q. What to do when there is no flow?
Ans:
- It is important to make sure that the pump is running, there is no blockage in the tubing provided in the system, and there is a continuous flow without stopping.
- Verify that there are no air bubbles in the pump heads causing pressure/flow changes or that there are no connection leaks.
- Disconnect the column and prime pump. flush system with 100% methanol or isopropanol. if the problem still persists contact the service engineer.
Q. What are the probable causes of no pressure/ or pressure lower than required?
Ans:
- Leakage of system
- Mobile phase flow interrupted/obstructed.
- Air trapped in the pump head
- Leakage at column inlet end fitting
- Air trapped elsewhere in the system
- Faulty check valve.
- Faulty pump seals.
Q. What to do when pressure is higher than usual?
Ans:
- Remove the guard column and analytical column from the system.
- Replace with unions and larger tubing to reconnect the injector to the detector.
- Run pump at 2 to 5 ml/min.
- Remove the guard column (if present) and check the pressure.
- If required, change the guard column. Reverse and flush the analytical column while detached from the detector if it is clogged.
- Filter sample; use a 0.5-micrometer in-line filter; disconnect and backflush column; replace inlet frit.
Q. What are the causes of noisy baseline?
Ans:
- It may be due to air in the mobile phase, detector, or pump.
- Pump pulsation.
- incomplete mobile phase mixing.
- It may be due to the effect of temperature (column heated to a high degree, unheated detector)
- Other electronic equipment on the same line.
- Leak
Q. What are the causes of the negative peak in the HPLC Chromatogram?
Ans:
- The Refractive index of the solute is less than that of the mobile phase (RI detector).
- The composition of the solvent obtained from the sample and the composition of the mobile phase are vastly different.
- The mobile phase has more absorption than sample components to UV wavelength.
Q. What are the causes of the loss of resolution and its solution?
Ans:
| Cause | Remedies |
| Mobile phase contaminated (causing RT to change) | Prepare a new mobile phase and discard others |
| Obstructed guard or analytical column | Remove the guard column and attempt analysis, replace the guard if necessary, and reverse and flush. if the problem persists, the column may be fouled with strongly retained contaminants. use the appropriate restoration procedure according to the manufacturer’s manual. if the problem still persists inlet is probably plugged. change frit or replace column. |
Q. What are the possible causes of the spike baseline and their remedies?
Ans:
| Causes | Remedies |
| Bubble in the mobile phase | De-gas mobile phase; use back-pressure restrictor at detector outlet; ensure that all fittings are tight. |
| Column stored without caps | Store column tightly capped; flush reversed-phase column with de-gassed methanol. |
| Poor electric connection, loose wiring | Clean and tighten detector leads, and check the wiring. |
Q. What are the causes of peak tailing?
Ans: Peak tailing may be due to the following possible causes:
- Sample reacting with active sites
- Wrong mobile phase pH
- Wrong column type
- The small(uneven) void at the column inlet
- Wrong injection solvent
- Void formation at the column head
- Column problems
- Guard problems.
Q. What are the possible causes of the broad peak in HPLC and remedies?
Ans:
| Causes | Remedies |
| Analyte elutes early due to sample overload | Dilute sample 1:10 in ratio and re-inject |
| Detector cell volume too large | Use the smallest possible cell volume consistent with sensitivity needs |
| The injection volume is too large | Decrease solvent strength of injection solvent to focus solute; inject smaller volume |
| Large extra column volume | Use low or zero dead volume end fittings and connectors; use the smallest possible diameter of connecting tubing |
| Viscosity too high of Mobile-phase solvent | Raise column temperature and switch to a solvent with lower viscosity |
| Peak dispersion in injector valve | The decreased injector sample loop size introduced air bubbles on the front and back of the sample in the loop. |
| Poor column efficiency | Use a lower flow rate, a higher column temperature, a mobile phase with lower viscosity, or a packing with smaller particle diameters. |
Q. What is the possible reason for peal doubling?
Ans: The possible reason for the peak doubling in the HPLC Chromatogram should be:
- BLocked frit
- Column overloaded
- Column void or channeling
- Injection solvent too strong
- The sample volume is too large
- Mobile phase composition change
- Mobile phase flow rate too low
- Cell volume or detector response time too large
- Buffer concentration too low
- Column temperature too low
Q. What are the courses of peak splitting?
Ans:
- The possible causes may be contamination on the guard or analytical column inlet.
- Partially blocked frit
- The small (uneven) void at the column inlet
- Sample solvent incompatible with the mobile phase.
- Column overloaded
- The sample volume is too large
Q. What are the types of baseline troubleshooting?
Ans:
- Noisy baseline
- cyclic
- Synchronous noise
- Asynchronous noise
- Drift
- Spike
- No peak
Note: HPLC interview questions with answers PDF Provided shortly
Frequently Asked Questions Based on HPLC
A negative peak means that there is less absorbance while the peak is passing through the detector than when the mobile phase has more absorbance than the analyte at the monitored wavelength.
RT is the amount of time it takes for the compound to pass through the column.
RRT is the comparison of the RT of one compound to another compound.
Peak tailing is measured by the tailing factor. It is calculated at 5% of the peak’s maximum height by dividing the distance from the front slope to the back slope of the peak by twice the distance from the peak’s center line to the front slope.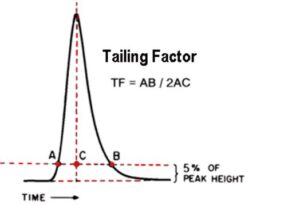
The sample is introduced into the mobile phase of the system using the needle so that it can be separated on the column. the needle wash is used to clean the needle after the injection.
Relative Response Factor (RRF) is an alternate method for the determination of the quantity of the impurity present in pharmaceutical products and the amount of the impurity can be calculated with the help of the peak area of the components.
De-Gasing is the term used to describe dissolved gases coming out of a solution. This phenomenon can also occur in a system where a rough surface produces nucleation sites for bubble formation. in analysis, the problems produced by bubble formation can largely be prevented, by De-Gassing the mobile phase.
The limit of detection (LOD) and limit of quantification (LOQ) are two important performance characteristics in method validation. The minimum concentration of an analyte that can be accurately measured by an analytical method is referred to as the LOD or LOQ.
The process of measuring the area under a chromatographic peak is known as chromatographic peak integration. The measurement uses the integral method, which involves cutting the peak into a lot of rectangles and adding them up to get an idea of how much space is underneath the peak.
Minimum area, tailing, and Fronting.

Naresh Bhakar is the Founder and Author at Pharmaguddu.com, bringing his extensive expertise in the field of pharmaceuticals to readers worldwide. He has experience in Pharma manufacturing and has worked with top Pharmaceuticals. He has rich knowledge and provides valuable insights and data through his articles and content on Pharmaguddu.com. For further inquiries or collaborations, please don’t hesitate to reach out via email at [email protected].