GMP & Pharmaceutical Manufacturing Expert
✓ Reviewed by: Pankaj Sharma - Quality Control Specialist
Reviewed for Quality Control accuracy, laboratory practices, analytical methods, and technical relevance
📅 Last Updated: February 16, 2024
CAPA stands for Corrective Action and Preventive Action. It is a tool used in quality management systems in the pharmaceutical industry. The purpose of corrective and preventive action is to analyze, identify problems, and implement appropriate measures to prevent their recurrence.
CAPA meanings/ Definition
Corrective action is the action taken to eliminate the causes of an existing problem, nonconformity, defect, or other objectionable events to prevent recurrence.
Preventive action is the action taken to eliminate the cause of potential nonconformity, defect, or other undesirable events to prevent their occurrence.
NOTE: Corrective action is taken to prevent a recurrence as defined in [ISO 9000:2005, clause 3.6.5], whereas preventive action is taken to prevent the occurrence.
Nonconformity refers to the failure to carry out a specific requirement.
Non-conformance refers to either the misinterpretation of written instructions leading to market or customer complaints, or the failure of equipment or the quality management system.
Related: Nonconformity during the audit and their types
Initiation process of Corrective and Preventive Action:
- Submit the source of information documents to the QA Head by the concerned department.
- Based on the source of information or risk factor, the QA Head and the concerned department will decide whether it is necessary or not.
- QA initiates the form along with its identification or reference number for further investigation.
- QA numbers it for identification in the following manner: (CAPA/DEPT.CODE/serial no./last two digits of a calendar year).
- The concerned department Head fills out the form with its initiation date and proposed completion date. Select the department by marking ✓ in the appropriate box. Mark ✓ on the affected site and select “not applicable” if the system is not affected. Write the description according to the source of information, along with corrective and preventive action. The concerned department head shall fill in their name with a signature and date.
- After filling out the form, the concerned department Head shall send it to QA for review.
CAPA as per ICH Q10?
A structured approach to identify the process should be used to determine the root cause.
13 Steps for CAPA in Pharmaceutical Industries:
The basic steps involved in applying effective Corrective and Preventive Action to improve the required documentation are as follows:
1. identification
Identification is the initial step in the corrective and preventive process, which involves defining a specific problem and its source of information. There is a requirement to document the available evidence for further use throughout the investigation process.
2. initiation of CAPA
CAPA shall be initiated after obtaining a source of information, which may be internal (e.g., internal audit) or external (e.g., customer complaint) with the CAPA source. It includes change control, deviation, incident report, product recall, man, machine, process method, materials, and environmental factors.
Related: Deviation, and Its types
3. CAPA Review
QA Head review for CAPA to determine what action has to be taken.
4. CAPA Acceptance or Rejection
Acceptance: If accepted, proceed with the initiation process of other Corrective and Preventive Actions by assigning a unique identification number for tracking and reference throughout the investigation.
Rejection: Document the reasoning (विचार ) and rationale (तर्क ) in case the issue repeats.
5. Evaluation and action
CAPA Evaluation is conducted to determine the nature and size of the problem, its impact, the associated risk, and the required level of action. For instance, it assesses the impact on product quality, safety, functionality, and customer needs. Additionally, assessing the risk is crucial for immediate remedial action, accompanied by a remedial action form.
6. The Process of Investigation
To ensure proper direction of an investigation, a written procedure should be conducted. The process should include designing the action strategy (writing steps involved in the analysis of root causes) and considering various factors such as machine, material, personnel, process, training, software, and other external factors.
7. Analysis or Determination of Root Cause
Root cause analysis is done by using the right analytical tools to analyze a problem. An accurate analysis leads to an effective action plan. The following methods can achieve this: Fishbone/Affinity (Ishikawa diagram), Five Whys, Fault tree analysis, and 5 M analysis.
Ishikawa or fishbone Diagram: It is used to analyze the root cause of a problem. Here is an example of a problem to investigate: “Tablet diameter out of specification.“
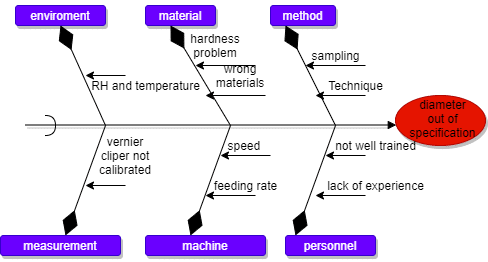
8. Develop an Action Plan
An action plan must be developed to prevent the future recurrence of the same problem. This assigns responsibility to the team to correct the problem and prevent its recurrence. If you are required to make changes in processes, documents, procedures, and other system-related modifications, including employee training, an action plan form should be included for CAPA. A case example is given below to develop the “5 WHYs” in your action plan.
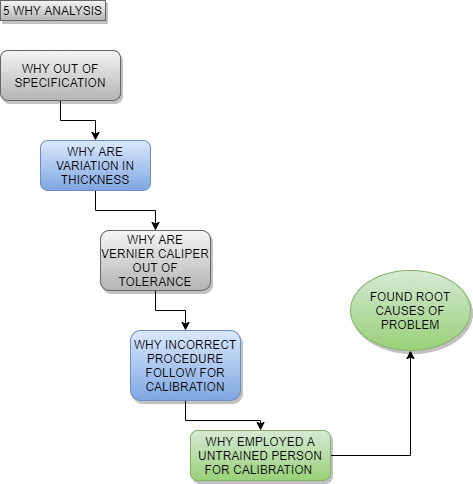
To analyze the 5 Whys, it is not simply asking five “why” questions, but each question must be connected to the next. In this scenario, the root cause of the out-of-specification issue might be the involvement of an untrained person in the process.
9. Completeness of the Action Plan
After creating the action plan, assign roles and responsibilities for plan execution. Create a task list for implementing it. The list may include:
- Update procedure.
- Add an inspection step.
- Re-design a process.
- Implementation of additional training for key staff.
- Switch material.
- Modify manufacturing /storage environments.
10. Verification, Effectiveness, and Validation of CAPA:
Ensure that the corrective and preventive action is effective and does not adversely affect the end products. Verify and review the data associated with it. Also, verify that a verification and validation protocol is established alongside this. Additionally, review the effectiveness of corrective and preventive action by ensuring that the data is reviewed to identify any similar problems.
11. implementation of Changes:
This step ensures that the control documents are thoroughly reviewed and approved in case any changes are made.
12. Management Review on CAPA
The Department Head shall certify that CAPA is implemented according to the process. QA shall verify it, along with the supporting documents, and provide certification. A copy of it shall be handed over to the concerned department Head. Finally, the QA Head and the QMS team will review CAPA to ensure that the process is followed correctly at all stages.
13. CAPA Closing
After completing all follow-ups and implementing corrective and preventive actions, the document should be dated and signed by an authorized department head.
Deviation and their Co-relationship with CAPA?
Deviation alone is not a solution to the problem. It can describe an issue, but to prevent its recurrence, Corrective and Preventive Actions (CAPA) are taken based on the nature of the problem. These actions help identify the root causes through investigation. The management of deviations must be recorded for future trend analysis.
The link between Complaint and CAPA?
Complain only triggers the CAPA. Remember, every complaint does not belong to CAPA.

Naresh Bhakar is the Founder and Author at Pharmaguddu.com, bringing his extensive expertise in the field of pharmaceuticals to readers worldwide. He has experience in Pharma manufacturing and has worked with top Pharmaceuticals. He has rich knowledge and provides valuable insights and data through his articles and content on Pharmaguddu.com. For further inquiries or collaborations, please don’t hesitate to reach out via email at [email protected].