GMP & Pharmaceutical Manufacturing Expert
✓ Reviewed by: Pankaj Sharma - Quality Control Specialist
Reviewed for Quality Control accuracy, laboratory practices, analytical methods, and technical relevance
📅 Last Updated: July 27, 2023
A cleaning Validation Master Plan (cVMP) is a very important document that is needed to be in place during the cleaning validation process. Below read the all content that helps to prepare a well-documented validation plan.
PROTOCOL CONTENTS
| S. No. | SECTION TITLE |
| 1.0 | APPROVAL |
| 2.0 | INTRODUCTION |
| 3.0 | PURPOSE |
| 4.0 | SCOPE |
| 5.0 | RESPONSIBILITIES |
| 6.0 | STRATEGY |
| 6.1 | PRODUCT / EQUIPMENT BRACKETING |
| 6.2 | CLEANING VERIFICATION |
| 6.3 | CLEANING TECHNIQUES |
| 6.4 | WATER QUALITY |
| 6.5 | ANCILLARY EQUIPMENT |
| 6.6 | EVALUATION OF CLEANING PROCEDURE |
| 7.0 | ACCEPTANCE CRITERIA |
| 7.1 | VISUAL INSPECTION |
| 7.2 | ACTIVE RESIDUE |
| 7.3 | MICROBIAL TEST |
| 7.4 | HOLD TIME STUDY |
| 8.0 | PROTOCOL |
| 9.0 | ANALYTICAL METHOD DEVELOPMENT AND VALIDATION |
| 10.0 | CHANGE CONTROL / REVALIDATION |
| 11.0 | DEFINITIONS AND REFERENCE |
1.0 APPROVAL
2.0 INTRODUCTION
2.1 Name of “COMPANY” is engaged in the manufacturing of the following dosage forms:
- Tablets ( Cephalosporin )
- Capsules
- Dry Powder Syrup ( Cephalosporin )
- Small-volume parenteral ( Cephalosporin )
- Others
2.2 This cleaning validation master plan is designed to demonstrate the approach of “COMPANY NAME” for cleaning validation to meet the current National and International regulatory guidelines.
2.3 At “COMPANY NAME” the cleaning procedure is manual for each piece of equipment.
2.4 The cleaning validation master is designed for instructions for planning, executing, and successfully completing the cleaning validation program. As the cleaning procedure is manual personnel validation will be performed during the cleaning validation.
3.0 PURPOSE
3.1 The cleaning validation master plan will be like a big umbrella guide for all the cleaning and steps we follow to make sure all the equipment used in “COMPANY NAME” are cleaned properly. This helps prevent any contamination that could change the safety, strength, purity, or quality of the medicine.
4.0 SCOPE
4.1 This cleaning validation master plan is applicable to the solid Oral dosage forms, Tablets, Capsules, and Powder Injectables manufactured at “COMPANY NAME”.
4.2 On introduction of new equipment/product it will be evaluated with the guideline provided in the cleaning validation master plan for determination of the requirement of cleaning validation.
5.0 RESPONSIBILITIES
5.1 Quality Assurance department will be responsible for:
- Executing and reviewing the Cleaning Validation Protocol.
- Coordinating with the production team for the sample collection as specified in the protocol.
- Ensuring that the equipment/ Machine is cleaned as per the respective Protocol.
- Swab/rinse sampling of equipment for chemical analysis.
- To prepare, review, and approve the Cleaning Validation Report.
5.2 Quality Control department will be responsible for:
- Reviewing the Cleaning Validation Protocol and report.
- Development of a validated analytical method of sufficient sensitivity as mentioned in the cVMP.
- Swab sampling for microbiological analysis.
- Analyzing the samples that is taken during the execution of the validation protocol.
5.3 Manufacturing department will be responsible for:
- Reviewing the Cleaning Validation Protocol and report.
- Provide training regarding the cleaning of equipment to the person who is involved in cleaning process.
- Executing the Cleaning Validation Programme.
6.0 STRATEGY (VALIDATION APPROACH)
6.1 PRODUCT / EQUIPMENT BRACKETING
- Due to the complexity of manufacturing multiple products and multi-product use of the same equipment, a bracketing approach shall be applied for cleaning validation based on scientific rationale. The low dose and the most insoluble drug product with the highest Batch Size will be selected for the cleaning validation purpose. In this approach cleaning procedure for each product will not be validated individually, however, it will provide a high degree of assurance of cleaning to prevent contamination that would alter the safety, identity, strength, purity, and/ or quality of the drug product to meet established specifications.
- Trains of equipment will be identified based on equipment capacity and batch size. The equipment with the largest and smallest capacity will be considered for cleaning validation/verification. If the equipment has various capacities but has similar design and operating principles, and the cleaning procedures are the same, perform cleaning validation on any of the equipment sizes. The following criteria shall be taken into consideration for the selection of products/equipment.
- Active Pharmaceutical Ingredient’s solubility (Difficult to clean and high dose) (Refer Annex-1A, 1B,1C,1D,1E)
- Low dose drug product (Refer Annex-1A, 1B,1C,1D,1E) Equipment train and its capacity (Refer Annex-2A, 2B,2C,2D,2E)
- Based upon an evaluation of the above criteria product matrix has been developed and the ‘worst case’ product will be used for the cleaning validation program. (Selection of product is presented in Annex –1).
- Change in cleaning agent in the cleaning procedure shall require revalidation, change in equipment that does not fit into the established equipment train will require revalidation.
6.2 CLEANING VERIFICATION:
6.2.1 Cleaning verification approach will be applied for the unique condition where the product is manufactured for the first time is not fitting / does not exist in the established product/equipment matrix until the cleaning procedure has been validated or it is one time activity/event the product will not be manufactured in the same manner on the commercial scale equipment. Equipment (Refer Annex-4).
6.2.2 Cleaning verification approach will be also applied while introducing/ replacing any equipment on the train, after appropriate evaluation. On the basis of evaluation, verification will be performed if required for particular equipment until cleaning procedure has been validated.
6.2.3 Cleaning verification will establish/demonstrate the proper removal of target product residue, approved detergent, and microbial load by which it will not alter the safety, identity, strength, purity, and/ or quality of subsequent drug products being manufactured on the same equipment.
6.2.4 Following criteria shall be adopted : Choose where to swab based on experience for hard-to-clean equipment spots or past worst-case samples.
6.2.5 Method adopted shall be qualified for the sensitivity and swab recovery.
6.3 CLEANING TECHNIQUES
The equipment used in the tablet department is cleaned with common cleaning technology. It consists of routine manual cleaning with raw water, followed by manual removal of adhered powder using a nylon brush, and then the surfaces are rinsed with raw water followed by hot Purified water. Dry by wiping with clean lint-free cloth or using filtered compressed air.
6.4 WATER QUALITY
Water to be used for the cleaning of manufacturing equipment is Raw water (Potable water) at room temperature and Purified water at a temperature not less than 45 ° C.
6.5 ANCILLARY EQUIPMENT
Use ancillary equipment with the main equipment in the equipment train. This extra equipment helps in making products by transferring materials, holding ingredients, and preparing solutions for granulation. Some examples of ancillary equipment are solution tanks, agitator assembly, and scoops. set up a cleaning validation for this ancillary equipment.
6.6 EVALUATION OF CLEANING PROCEDURES
6.6.1 To evaluate the cleaning procedure and to give evidence of cleaning effectiveness following methods will be used:
(1) Visual inspection,
(2) Swab sample
(3) Rinse sample ( where it is applicable)
6.6.2 Method adopted for the analysis of the sample shall be suitably validated as mentioned in section 9.0 of this master plan.
6.6.3 Visual inspection will be performed upon completion of the cleaning procedure. Swab sampling will be performed only after satisfactory visual inspection has been completed. Unsatisfactory visual examination, swab, or rinsate sample results attributed to the cleaning procedure, will require that the following steps be taken.
6.6.3.1 The failing cleaning procedure will be revised. perform and write about the investigation in the validation summary report, also validate the revised cleaning procedure.
6.6.3.2 Unsatisfactory visual inspection results will require re-cleaning using the existing cleaning procedure. Proper cleaning must be verified prior to the use of the equipment for manufacturing. Swab or rinse samples may be used to verify the re-cleaning.
6.6.3.3 Equipment, which after the cleaning, has passed visual inspection, but has subsequently failed to meet the analytical or microbiological acceptance criteria for swab or rinsate sample will require re-cleaning using the existing cleaning procedure. The Data collected during the re-cleaning process cannot be utilized to confirm the effectiveness of the cleaning procedure. In cases where equipment used in product manufacturing fails and leads to unsatisfactory analytical or microbiological results, it is important to investigate the impact before approving the release of the affected products.
6.6.3.4 Production department in consultation with Quality Assurance will initiate an investigation as to the causes of the unsatisfactory results.
6.6.3.5 Sampling and analytical methods will depend on the nature of residue and manufacturing equipment. The cleaning validation protocols will specify the sampling techniques and locations.
6.6.3.6 Rinse sample can be applied to equipment that can hold solvent/water, and is used between successive rinse steps to evaluate the effectiveness of the rinsing steps.
6.6.3.7 Swabbing is the preferred sampling technique since it is used for establishing the levels of residues of moderately or insoluble active ingredients, cleaning agents, and microbial bioburden. Swabbing of product contact surfaces of manufacturing equipment and utensils must include “ hard to clean” areas. Operator and supervisor’s experience as to the most difficult-to-clean locations must be taken into account. Most equipment will be swabbed in 3-5 locations, depending on the equipment size, accessibility, and complexity. Equipment with a small surface area, such as Deduster, will be swabbed in 1-3 locations. Instructions for swabbing will be specified in each validation protocol for each piece of equipment. The procedure will specify the following.
- Type of swab.
- Type and amount of solvent used to extract the swab.
- Swabbing technique.
- Dimension of equipment surface area to be swabbed (e.g. 10 cm x 10 cm).
- Holding time and conditions for swab samples prior to testing.
- Ensuring the Proper Holding Time and Conditions for Swab Samples Before Testing.
- Before conducting the tests, it is important to handle the swab samples correctly. The samples will be carefully placed in suitable containers, tightly sealed, and clearly labeled.
- Equipment identification / Location
- Product name and strength
- Batch number
- Sampled by, time and date of sampling.
7.0 ACCEPTANCE CRITERIA
The cleaning process is considered validated once it meets the acceptance criteria as per given in the protocol. The failure of individual sampling points will not necessarily mean that the cleaning method is inadequate. Each deviation will be investigated and based on the investigation, corrective actions will be taken that may require further follow-up or further validation.
7.1 Visual Inspection:
After the cleaning of the equipment, a visual inspection will be performed prior to another sampling. Therefore, a visual inspection will be part of the cleaning validation acceptance criteria for all equipment and utensils. The clean dry equipment must contain no visible residue.
7.2 Active Residue:
Calculation of active residue after cleaning will be based on product contact surface/area. This approach is based on the acceptable daily intake (ADI). ADI is defined as the amount of the drug product (mg/day), that a person can be exposed as a contaminant in another pharmaceutical product without experiencing any adverse health or pharmacological effects. Based on the above, the calculation for carryover is based on the assumption that only a fraction (1/1000) of the smallest daily dose of product “A” can be carried over to the maximum allowed daily dose of product “B” manufactured in the same equipment. The factor of 1/1000 was obtained based on; the oral drug products are pharmacologically inactive and safe at 1/1000 of their normally prescribed dosage; The following formula will be used for determining the residue level. (Refer to Annex-3 for the calculated limit for the active residue).
Read About: MACO and NOEL Calculation for Cleaning Validation
MACO = STD X SBS/
SF X LDD
Where,
MACO = Maximum allowable carryover
STD = Smallest therapeutic dose of any product manufactured in equipment train. (Product-A)
SBS = Smallest batch size of any product manufactured in equipment train. (Product-B)
SF = Safety factor 1000
LDD = Largest daily dose of any product manufactured in equipment train. (Product-B)
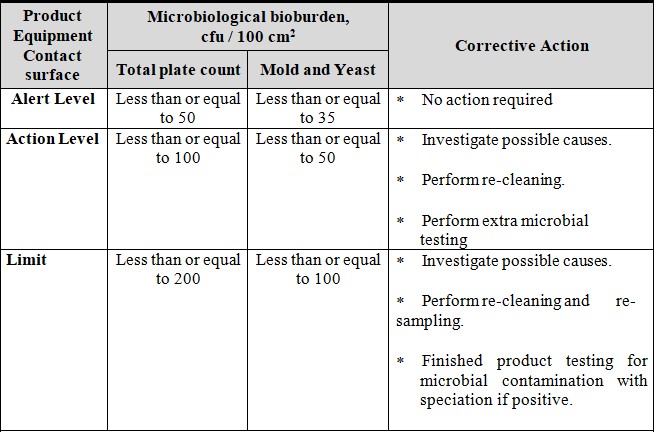
7.3 MICROBIAL TEST:
Swab samples are to be collected from the product contact surface area immediately after the completion of cleaning activities and after the specified hold time period for the total aerobic microbial count. The limits for the microbiological bioburden criteria for product contact surfaces are presented below:
7.4 HOLD TIME STUDY:
To establish the effectiveness of cleaning equipment will be kept idle for 72 hrs. in a dirty condition. To establish the expiry of cleaning in view of microbiology, equipment shall be kept idle after cleaning for 72 hrs., and microbiological swabs shall be taken and analyzed. This will be considered as worst case and microbial load should remain well within the limit.
Related: Protocol on Hold Time Study of Machine Parts
8.0 PROTOCOL
8.1 A cleaning validation protocol shall be developed for the ‘worst case’ product selected for the cleaning validation program. Following information but not limited to the following included in the cleaning validation protocol. For numbering of protocol refer to SOP:
8.1.1 Objective: A brief description of the purpose of the validation study.
8.1.2 Scope: This section must include the extent of the cleaning validation protocol.
8.1.3 Responsibilities: This section includes the different responsibilities for completing the cleaning validation program.
8.1.4 Protocol Signature Log: This document contains the signatures (specimens) of all individuals participating in the cleaning validation program. These signatures are recorded to accurately identify each person for future reference purposes.
8.1.5 Protocol training record: Training is to be provided to all personnel involved in the cleaning validation program for understanding and cGMP requirements.
8.1.6 Equipment Description and Cleaning Procedure: A brief description of the equipment used in the manufacturing of the selected product and the relevant SOP for equipment cleaning.
8.1.7 Sampling Plan and Sampling Procedure:
- A number of satisfactory cleaning validation runs shall be specified.
- The type and number of samples during each validation run shall be specified.
- In addition to visual inspection, the sampling plan shall include swab sampling of product contact surfaces.
- The location of product contact surface samples (swab samples) shall be referenced on the equipment drawing.
- The sampling plan shall include swabs for active residues and cleaning agents, rinse samples, wherever applicable, and microbial tests.
Each protocol will include test forms, including the given detail:
8.1.7.1 Equipment Identification.
8.1.7.2 Product name and strength.
8.1.7.3 Batch number.
8.1.7.4 Name of the person who performed the cleaning.
8.1.7.5 Name of the person who verified the cleaning.
8.1.7.6 Name of the person who performed the sampling.
8.1.7.7 Swab sample specific Location.
8.1.8 Analytical and microbiological test methods: This section will give references for the adopted analytical and microbiological test methods to analyze the samples. The analytical method must be validated for the residue levels, or bioburden as per the specification given in the protocol. Testing performed by “COMPANY NAME”. All test results must be calculated and reported to correspond with the predetermined acceptance criteria.
8.1.9 Acceptance Criteria: The criteria for accepting the cleaning validation will be as per the given protocol and determined according to the matrix of the product.
8.1.10 Deviation:
Any deviation taken during the execution of the protocol shall be documented in this section. Justification for the deviation will be authorized by Quality Assurance Manager and GM QA / QC.
8.1.11 Summary report:
After the completion of the cleaning validation program for the selected product, a final report will be generated for each protocol. The report shall contain a summary and discussion of validation testing and result including relevant supporting documents. Deviation from the cleaning validation protocol will be noted in the report. The summary report will be approved by Quality Assurance Manager and GM QA / QC.
9.0 ANALYTICAL METHOD DEVELOPMENT AND VALIDATION
9.1 The development and validation of analytical procedures for the purpose of analyzing cleaning validation samples requires the selection of appropriate tests.
9.2 The list of such tests, with their definition is given below:
9.2.1 LIMIT OF QUANTITATION:
The limit of quantitation for an individual analytical procedure refers to the smallest amount of a substance in a sample that can be accurately and precisely measured.
9.2.2 LIMIT OF DETECTION:
The limit of detection of an individual analytical procedure is the lowest amount of sample, which can be detected but not necessarily quantitated as an exact value.
9.2.3 LINEARITY:
The linearity of an analytical procedure is its ability (within a given range) to obtain test results that are directly proportional to the concentration of analyte in the sample at the LOQ level.
9.2.4 RECOVERY STUDY:
This study is to check the efficiency of the swab sampling procedure from the surface (Contact surface like SS, Teflon rubber, etc.). The study is performed by applying the known concentration of the standard solution on the surface at target and LOQ levels. The recovery study will be performed by different analysts to demonstrate the robustness of the analytical procedure.
10.0 CHANGE CONTROL / REVALIDATION :
Any changes to a parameter of a previously validated cleaning procedure must be evaluated by the Quality Assurance department to determine if additional cleaning validation is required. The following changes may affect the validated status of a cleaning procedure.
- New product
- Major product reformulation
- Equipment changes (size or type)
- Cleaning agent changes, or cleaning agent formulation changes
- Cleaning procedure changes
- Repeated cleaning failures
The list of changes that could potentially affect the validation status of a cleaning process is not all-inclusive, i.e. there could be other changes that would also require revalidation.
11 DEFINITION & REFERENCES:
11.1 DEFINITION
11.1.1 CLEANING VALIDATION:
Cleaning Validation is defined as “Establishing documented evidence that the cleaning process consistently provides a high degree of assurance that after cleaning, equipment and system are free from materials that would contaminate or adulterate subsequent product up to the predetermine acceptance criteria ”.
11.1.2 EQUIPMENT TRAIN: Equipment train is defined as a “Group of equipment in sequence, utilized for manufacturing particular group of product, considering the batch size and capacity of equipment”
11.2 REFERENCE:
11.2.1 Schedule – M
11.2.2 Code of federal regulation -211, subpart 211.67
11.2.3 I. R. Berry and D. Harpaz, eds. 1997, Validation of bulk pharmaceutical chemicals.
11.2.4 Cleaning Validation Master Plan 200.1, CARACO Pharmaceutical Laboratories Ltd
11.2.5 Addendum to Cleaning Validation Master Plan VMP01-05.06, CARACO Pharmaceutical Laboratories Ltd

Naresh Bhakar is the Founder and Author at Pharmaguddu.com, bringing his extensive expertise in the field of pharmaceuticals to readers worldwide. He has experience in Pharma manufacturing and has worked with top Pharmaceuticals. He has rich knowledge and provides valuable insights and data through his articles and content on Pharmaguddu.com. For further inquiries or collaborations, please don’t hesitate to reach out via email at [email protected].