Quality Control Specialist | Industry Experience
✓ Reviewed by: Pankaj Sharma - Quality Control Specialist
Reviewed for Quality Control accuracy, laboratory practices, analytical methods, and technical relevance
📅 Last Updated: February 20, 2023
Objective: To lay down procedures for validation of Analytical Procedures to be followed in the laboratory. The AMV (Analytical method Validation) aims to demonstrate that it is appropriate for the purpose.
Scope: This SOP is applicable in Analytical Research and Development Laboratory (ADL) for validation or verification of analytical procedures at the Pharmaceutical Quality control laboratory.
References: Method Validation of Analytical Procedures ICH, WHO TRS – Appendix-4 (QAS/16.671/Rev.1), Q2 (R1).
Procedure for Method Validation of Analytical Procedures:
Method Validation of analytical procedure is the process by which it is established, by laboratory studies, that the performance characteristics of the method meet the requirements for the intended analytical applications through certain accuracy and reliability standards.
Prepare a validation protocol and get the proper authorities to approve it. Before starting the Validation experiments, system suitability shall be done to determine the suitability of the Chromatographic system for the analysis as per the individual method. Theoretical Plates, capacity, resolution, and tailing factors must all be determined/calculated if necessary. When the System meets the system suitability parameters mentioned in the method, validation Experiments shall be started.
Bracketing standards shall be injected throughout the validation. The mean, standard deviation, and relative standard deviation shall be determined as per the individual method system suitability criteria.
Types of Analytical Parameters Used in Validations for (Assay):
- Specificity (Note: Specificity must be carried out using PDA Detector.)
- Forced Degradation Study
- Linearity
- Precision
- Accuracy
- Stability of Analytical Solution
- Robustness
- Filter Study
Types of Analytical Parameters Used in Validations for (Related substances)
- Category I All Parameters
- Limit of Detection (LOD)
- Limit of Quantitation (LOQ)
Types of Analytical Parameters Used in Verification
- Precision
- Linearity
- Specification
Data Elements Required For Validation & Verification:
Considering a variety of test methods (Assay, Related substances, etc.) it is only logical that different test methods require different validation schemes. The most common categories of methods for which validation data should be required. These categories are as follows:-
Category I: Analytical methods for Quantitation of major components of active ingredient (including Preservatives) in finished pharmaceutical products (Assay).
Category II: Analytical Procedures for assessing degradation components in final pharmaceutical products or impurities in bulk pharmacological ingredients These procedures include limit tests and quantitative assays. Analytical methods for determination of impurities in degradation compounds in finished pharmaceutical products. These methods include related substance tests.
Category III: Analytical procedures for determination of performance characteristics (e.g., dissolution, drug release, and others).
Category IV: Identification: For each category, different analytical information is needed. Listed in the Table are data elements that are normally required for each of these categories.
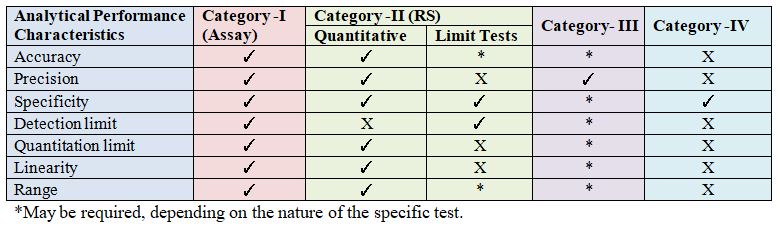
Analytical Parameters used in Validation of Analytical Procedures & Verification are elaborated below:
System Suitability:
System suitability is an integral part of many analytical procedures. System Suitability tests are based on the concept that the equipment, electronics, and analytical operations are samples to be analyzed and constitute an integral system that can be evaluated as such System suitability tests are used to verify that their solution and reproducibility of the chromatographic system are adequate for the analysis to be done.
The resolution is the function of column efficiency and it is specified to ensure that closely eluting compounds are resolved from each other. To establish the general resolving power of the system & to Ensure that internal standards are resolved from the drug. Column efficiency, tailing factor, theoretical plates, capacity factor, and relative standard deviation are the test Parameters to be established for the particular test procedures depending on the type of procedure being validated.
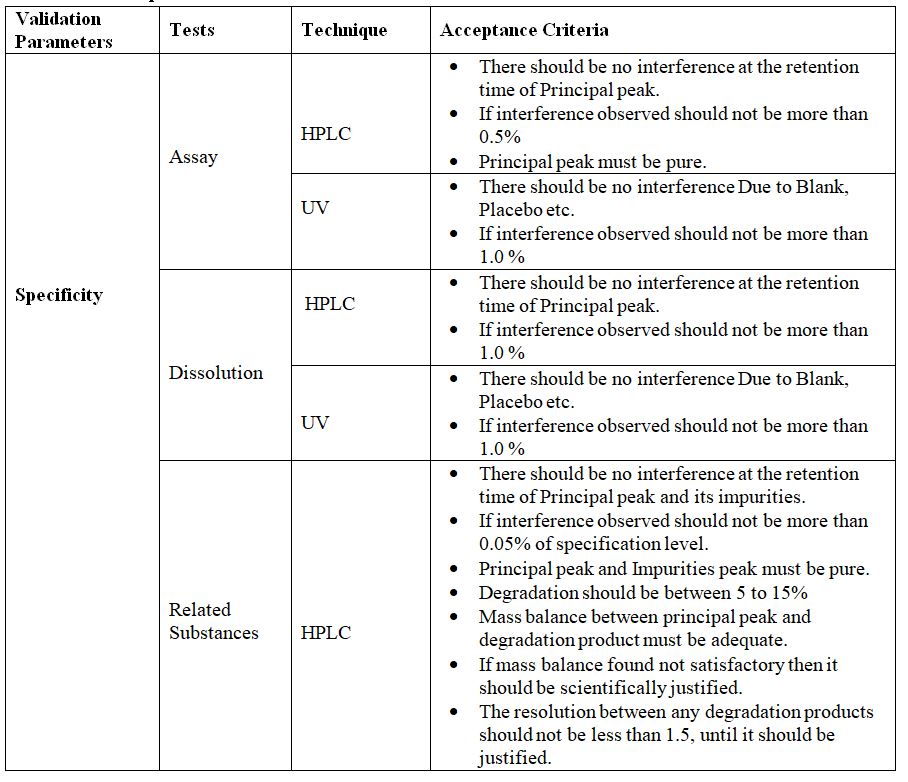
Specificity:
The term specifically refers to a method, which produces a response for only a single Analyte. The terms specificity and selectivity are often used interchangeably. Selectivity refers to a method, which provides responses for a number of chemical entities, which may or may not be distinguished. If the response is distinguished from all other responses the method is said to be selective.
The specificity of an analytical method is its ability to measure accurately and specifically, the analyte in the presence of components that may be expected to be present in the sample mass or matrix. It is often been expressed as the degree of inclination of test results obtained by analysis of samples containing added impurities, degradation products, related chemical compounds, or placebo ingredients When compared to test results from samples without added substances.
Procedure: In this study for related substances test individual impurities are injected and their retention times are determined and proved that the retention times are specific and there is no interference with each other. For assay studies limit level impurities are spiked in the product and the specificity of the test is proved by determining the assay which
Should be in Limit.
Acceptance Criteria:
1) RT should be distinct for different impurities.
2) Resolution between the two closest peaks should be as high as possible.
3) For Titrimetric assay the assay value should be within limit
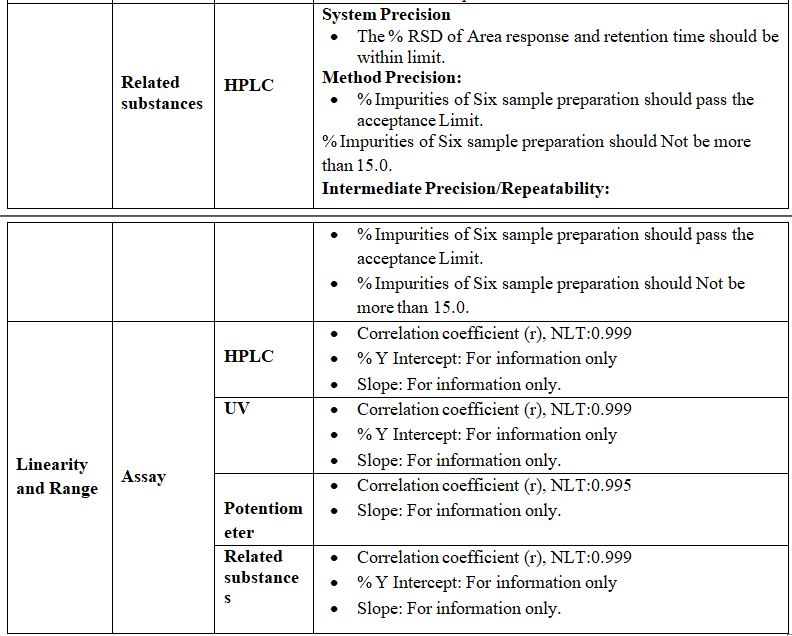
Linearity: (For Category I, II & III)
The linearity of an analytical method is its ability to put across test results that are directly or by a well-defined mathematical transformation, proportional to the concentration of Analyte in samples within a given range. Linearity is expressed in terms of the variance around the slope of the regression line, calculated according to an established mathematical relationship from test results obtained by the analysis of samples with Varying Concentrations of the analyte.
The linearity of an analytical method is determined by the mathematical treatment of test results obtained by analysis of samples with analyte concentrations across the claimed range of the method. The slope of the regression line and its variance provide a mathematical measure of linearity. Plotting the test results in Graphically as a function of analyte concentration is also acceptable.
Acceptance Criteria: The correlation coefficient should be NLT 0.995.
Forced Degradation Study:
In order to prove the specificity of the method, further degradation study was carried out On the Proposed method and were monitored for peak purity. The following degradation Conditions were used. It was observed that the percentage degradation was 5% to 20%.
Acid Degradation, Alkali Degradation, Peroxide Degradation, Thermal Degradation, UV Exposer Degradation, and Photostability Degradation.
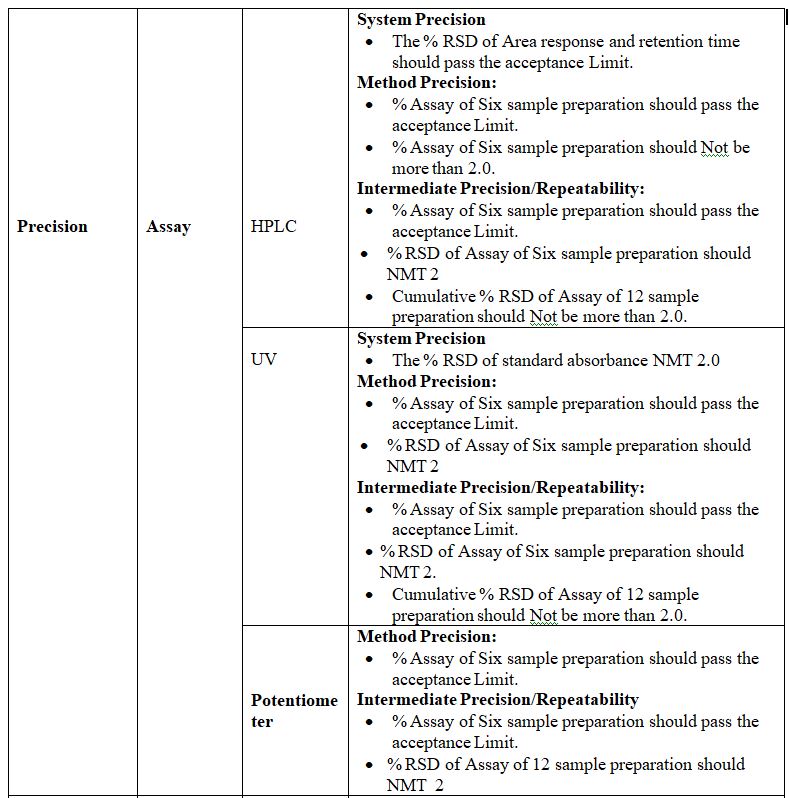
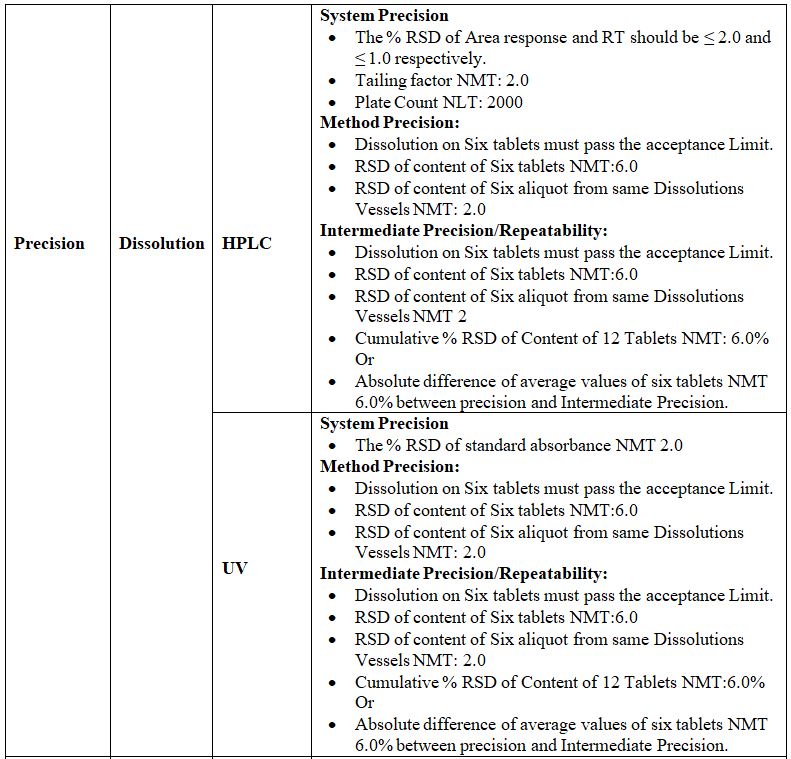
Precision:
Definition: The precision of an analytical method is the degree of agreement among individual test results when the procedure is applied repeatedly to multiple samplings of an analytical method usually expressed as standard deviation or Relative standard deviation. By assaying a sufficient number of aliquots of a homogenous sample to be able to calculate a standard deviation or relative standard Deviation, precision can be determined.
The precision is of three types:-
1. System Precision:
Definition: Replicate injections of standard preparation are compared to ascertaining precision tests, over a short interval of time on the same equipment and analyst.
Procedure: In this study standard solution or system suitability solution test is Injected in six replicates and the %RSD is established.
Acceptance Criteria:
1) Resolution between two peaks should be maximum or as specified in the method.
2) %RSD for RT should be NMT 1, 2, or as specified in the method.
3) %RSD for Area should be NMT 2,5,15 or as specified in the method.
2. Method Precision:
Definition: The precision of an analytical method is the degree of agreement among individual test results when the method is applied repeatedly to multiple samplings of a homogenous sample. It is a measure of either degree of reproducibility or repeatability of the method. If known impurities are not detected in the test sample under consideration for validation, method precision shall be justified by spiking the known concentration of impurities.
Procedure: In this study, six different test samples prepared separately shall be injected and the %RSD of these six results is determined.
Acceptance Criteria: The limit is between 2.0% to 10.0% for related substances tests and 2.0% for assay tests. This depends on the test being carried out.
3. Intermediate Precision:
Definition: Intermediate Precision (Ruggedness) of an analytical method is the degree of repeatability of test results obtained by the analysis of the same samples by/different analysts/ different days/different instruments/columns etc.
Procedure: In this study, six different test samples prepared separately are injected and the %RSD of these six results is determined by changing the different physical Parameters like different analysts/different days/different instruments/Columns, etc.
Without changing any analytical parameter. Also, the %RSD of 12 results obtained from different sets are determined.
Acceptance Criteria: The limit is between 2.0% to 10.0% for related substances tests and 2.0% for assay tests. This depends on the test being carried out.
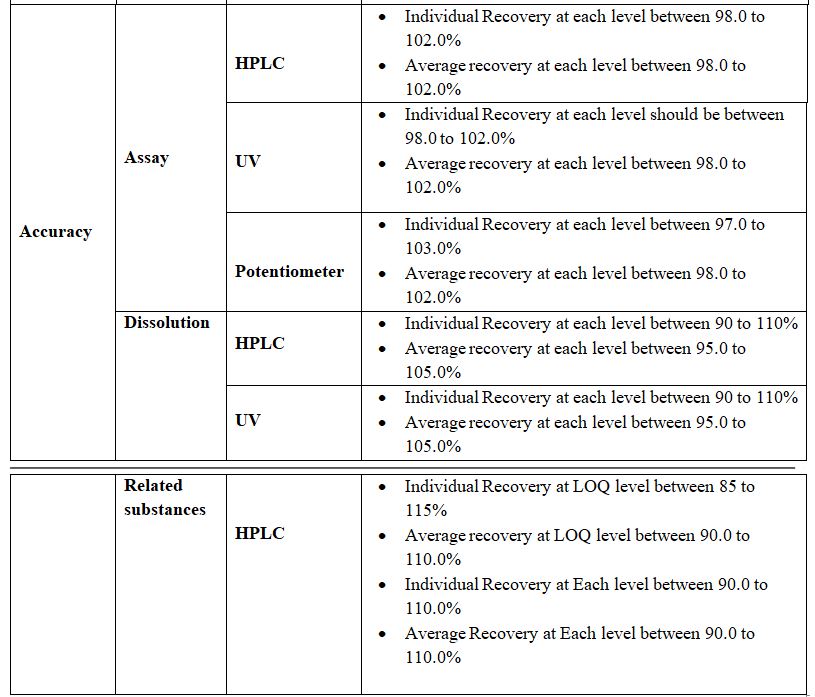
Accuracy:
Definition: A procedure is deemed accurate if, on average, the method provides the true answer. It is also defined as the closeness of test results obtained by that method to the true value. It may be expressed as percent recovery by the assay of known added amounts of analyte/impurities. Accuracy is a measure of the exactness of the analytical method. Design appropriate experiments and generate data. Study and assess whether it is in supports the accuracy of the method.
Procedure: This study is performed on LOQ, 80.0%, 100.0%, and 120.0% impurity Level concentration in related substances test and 80%, 100%, and 120% test level concentration for assay test. The amount of added or spiked analyte/impurity depends on the limit of particular impurities in the product, e.g. if the limit of a particular impurity is NMT 0.10% then we have to spike the impurity to 0.1% level with respect To the test concentration for 100% level.
Acceptance Criteria: The recovery should be between 80.0% to 120.0% for levels of 80%, 100%, 120%, and 70% to 130% for LOQ. The %RSD of recovery for three replicate injections should be between 2.0% to 10.0% for related substances test and 2.0% for assay tests
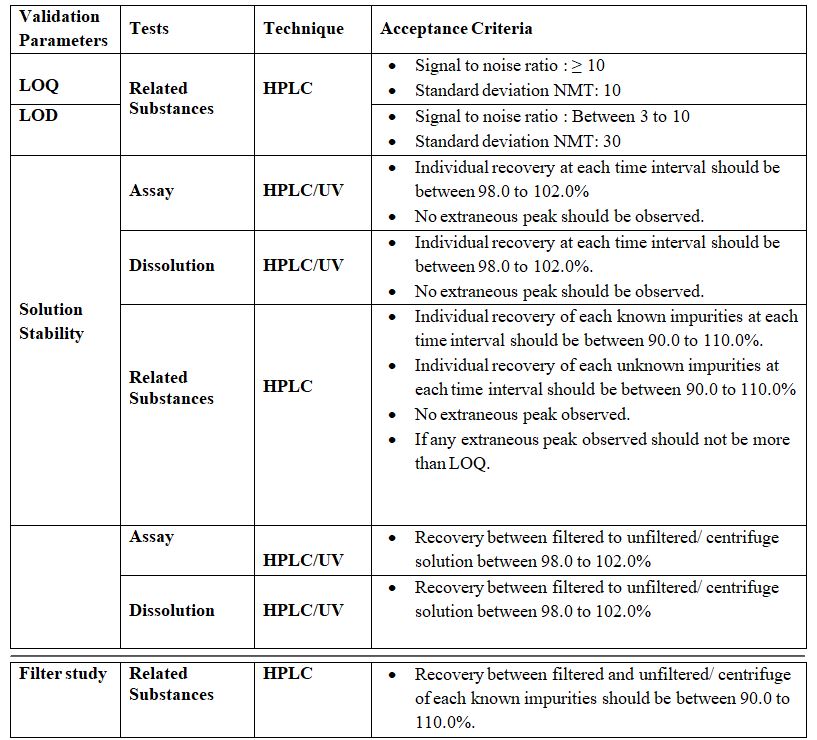
Limit of Detection (LOD):
The ability to detect, the lowest or smallest concentration of analyte in a sample is expressed as the limit of detection (LOD). Limit tests substantiate that the analyte concentration is above or below a certain limit. For instrumental methods, the lowest concentration of impurities which gives a recognizable peak is the limit of detection.
Based on visual evaluation:
In the case of non-instrumental analytical methods and also with instrumental methods, the limit of detection (LOD) is determined by the analysis of samples with known Concentrations of analyte and by establishing the minimum level at which the analyte Can be reliably detected. In case, if repeatability (%RSD) is well within the acceptance limit, however, peak shape is not proper this level should be considered as the limit of detection.
For instrumental methods, different techniques are used such as keeping signal to noise ratio of 2: 1 or 3: 1. Alternatively, the magnitude of the analytical background response Is measured by analyzing a number of blank samples and calculating the standard Deviation of this response. Standard deviation, multiplied by a factor, usually 2 or 3 provides An estimate of the limit of detection. Any one of the concepts can be used to ascertain LOD.
Procedure: In this study, the impurities are serially injected by lowering their Concentrations and their LOD values are determined based on the %RSD criteria.
Acceptance Criteria: The lowest concentration of impurities which gives a
recognizable peak is the limit of detection
Limit of Quantitation (LOQ):
The Quantitation limit of an individual analytical procedure is the lowest amount of the analyte in a sample, which can be quantitatively determined with suitable precision and accuracy. The quantitation limit is a parameter of quantitation assays for low levels of compounds in sample matrices and is used particularly for the determination of impurities and/or degradation products. It may be determined by the analysis of samples with the known concentrations of the analyte and by establishing the minimum level at which the analyte can be quantified with acceptable accuracy and precision. In this study, the impurities are serially injected by lowering their concentrations and their LOQ values are determined.
Based on visual evaluation:
For non-instrumental analytical methods and also with instrumental methods, the limit of quantitation is determined/ by the analysis of samples with known concentrations of analyte and by establishing the minimum level at which the analyte can be detected With acceptable accuracy and precision.
Procedure: In this study, the impurities are serially injected by lowering their Concentrations and their LOQ values are determined based on the % RSD criteria.
Acceptance Criteria: The lowest concentration of the Impurity which has a %RSD less than the limit given in the individual protocol is the limit of Quantitation.
For instrumental procedures, the same method may be used as for non-instrumental. The quantitation limit is shown to be sufficiently low by the analysis of samples with Shown concentrations of analyte above and below the quantitation limit.
Determination of signal-to-noise ratio is performed by comparing measured signals from samples with the known concentration of analyte with those of blank samples and by establishing the minimum concentration at which the analyte can be reliably Quantified. A typical signal-to-noise ratio is 10:1 Based on the standard deviation of response and the slope the quantitation limit can be expressed as :
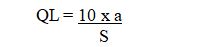
Where ‘S’ is the slope of the calibration curve and ‘a’ is the standard deviation of the response
Stability of Analytical Solution:
The sample solution was prepared as per the proposed Method & measure the area/absorbance at initial & different time intervals at Room temperature & 8°C.
Robustness:
It is the ability of the procedure to provide analytical results of acceptable accuracy And precision under a variety of conditions. The results from separate samples are Influenced by changes in the operational or environmental conditions. Robustness should be considered during the development phase and should show the reliability of an analysis when deliberate variations are made in method parameters.
Procedure: In this study, we make small but deliberate variations in analytical test parameters are made. For e.g. validation in pH, Mobile phase composition, flow rate, Oven temperature, etc. are made. The same batch material is analyzed by making variations in analytical test parameters.
Acceptance Criteria: The %variance of results from the standard condition result Should be NMT 10.0%. The parameters vary from method to method.
Solution Stability:
Determine the % difference of peak areas of consecutive injection in the sample solution and standard solution at Solution stability shall be determined by analyzing standard and sample solutions at an initial stage and at various periodic intervals up to about 24 hours. Area response of initial time (Zero hours) shall be compared with periodic interval results. The study shall be stopped in case of two consecutive failures of standard and sample solution at each interval. Stability in the Analytical solution test is to be validated for the Assay only.
Prepare standard solution and sample solution as per methodology, inject the standard & sample solution as per the below table with specific intervals of time
(0,4,8,12,16,20, and 24hrs.)
Procedure: In this study standard solution or system suitability solution test is injected in six replicates and the %RSD is established. The system suitability parameters vary From method to method.
Acceptance Criteria:
1) Resolution between two peaks should be maximum or as specified in the method.
2) %RSD for RT should be NMT 1 or 2 or as specified in the method.
3) %RSD for Area should be NMT 2,5, and 15 or as specified in the method
Filter Interference Study
Filter study test represents an integral part of the method and is used to ensure the effectiveness of the filter on the solution. Filter study test is to be validated for Analysis. Prepare Blank solution, Standard solution & Sample solution as per the methodology. The same Sample Solution is to be filtered through, a 0.45µ Nylon Filter, 0.45µ PVDF syringe filter, 0.45µ PTFE syringe filter & Whatman Filter No. 1, and one portion is to be centrifuged and injected.
Acceptance Criteria:
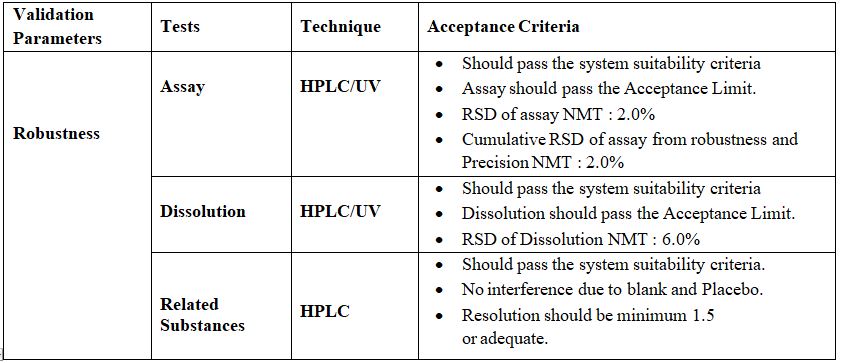
Analytical methods for Verification of Pharmacopoeia Products:
These methods include Verification tests of Pharmacopoeia Products. The following parameters are performed for verification.
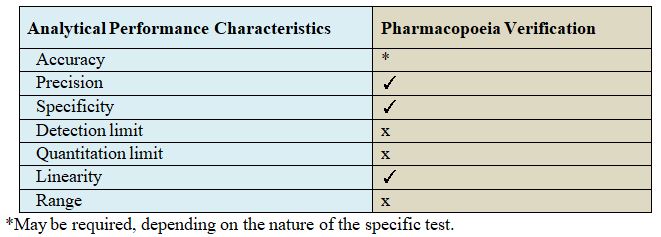
Matrix Plan for validation:
Matrixing designs can be applied to strengths with identical or closely related formulations.
Examples include but are not limited to
(1) Capsules of different strengths made with different fill plug sizes
(2) Tablets of different strengths manufactured by compressing varying amounts and
(3) Oral solutions of different strengths with formulations that differ Only in minor Excipients (e.g., Colorants or flavorings).
Revalidation Criteria: Revalidation shall be necessary for the following circumstances:
Change the Label Claim (API & Preservative) of the Product. Changes in the Synthesis of the drug substances. Changes in the composition of the finished product. Changes in the analytical procedure.
Analytical Method Transfer Protocol & Report shall be prepared as Annexure-I
All the validation activities shall be carried out as per SOP and pre-approved method validation
The protocol shall be used for analysis as per Annexure-I. Primarily method validation protocol issued as per Annexure-I
Abbreviations:
SOP: Standard Operating Procedure
AD: Analytical Development
LOQ : Limit of Quantitation
RSD: Relative standard deviation
LOD: Limit of detection
SST: System suitability test
RRT: Relative retention time
HPLC: High-Pressure Liquid Chromatography
Conclusion:
Method Validation of Analytical Procedures is applicable in the quality control lab and has a significant role in the verification of system suitability, specificity, and linearity.

Reviewed by: Pankaj Sharma — Quality Control Specialist
Pankaj Sharma is a pharmaceutical QC professional who reviews our technical content for GMP accuracy, laboratory practices, and regulatory relevance.