GMP & Pharmaceutical Manufacturing Expert
✓ Reviewed by: Pankaj Sharma - Quality Control Specialist
Reviewed for Quality Control accuracy, laboratory practices, analytical methods, and technical relevance
📅 Last Updated: September 8, 2024
As a pharmaceutical professional, we generally face various problems during analytical processes in our work such as HPLC, GC, IR, UV, Mass Spectroscopy, and many others. These instrumental methods are essential for the analysis of drug substances and drug products to ensure their quality, safety, and efficacy.

21 Top HPLC, GC, IR, UV, Mass Spectroscopy Questions and Answers:
In this article, we will focus on some common interview questions related to these analytical techniques.
Q.1 What is HPLC?
Ans: The separation, identification, and quantification of components in a mixture is done using a technique called high-performance liquid chromatography (HPLC). A sample is driven through a column containing small particles and a mobile phase that carries the sample through the column. As it goes through the column, different components within the sample will interact with the particles and mobile phase at different rates promoting their separation. These separated components are then detected as peaks on a chromatogram.
Q.2 How does HPLC differ from GC?
Ans: Gas chromatography also known as GC is an analytical method similar to HPLC but which uses gaseous rather than liquid as its mobile phase. This facilitates for vaporizable compounds that cannot be separated using HPLC. Generally speaking, GC requires that samples be in gaseous states while HPLC can detect both solid and liquid samples.
Q.3 What is IR spectroscopy?
Ans: In easy form, Infrared (IR) spectroscopy is a way of gauging how light molecules absorb infrared radiation. Light gets absorbed at different wavelengths for various functional groups within molecules which help to identify what these specific compounds are. For the identification of compounds IR spectroscopy is typically used as a complementary technique to HPLC and GC.
Q.4 What is UV spectroscopy?
Ans: Ultraviolet (UV) spectroscopy is a method of measurement of absorption of ultraviolet radiation by substances. It is commonly used for conjugated double bond analyses, and also quantitative analysis can be done. UV spectroscopy is frequently applied in pharmaceutical laboratories to ascertain levels of active ingredients in medicinal drugs.
Q.5 What is mass spectrometry?
Ans: Mass spectrometry is a procedure or process that determines mass to charge ratio of ions. In this method, the sample gets ionized then the ions are separated based on their mass-to-charge ratio before they are detected and finally, there is a resulting mass spectrum. Quantification, compound identification as well as structural analysis are examples where this technique has been useful.
Q.6 What are some acceptance criteria for dissolution as per USP?
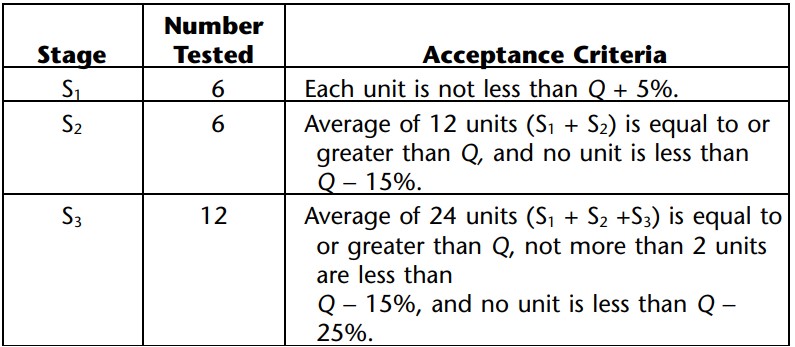
Ans: If we talk about acceptance criteria for dissolution as per USP, The United States Pharmacopeia (USP) has its own set standards for the quality of drug products, including their dissolution profiles. Some common acceptance criteria for dissolution as per USP include a minimum of Q+5% for the S1 stage. See the table below:
Immediate-release tablets:
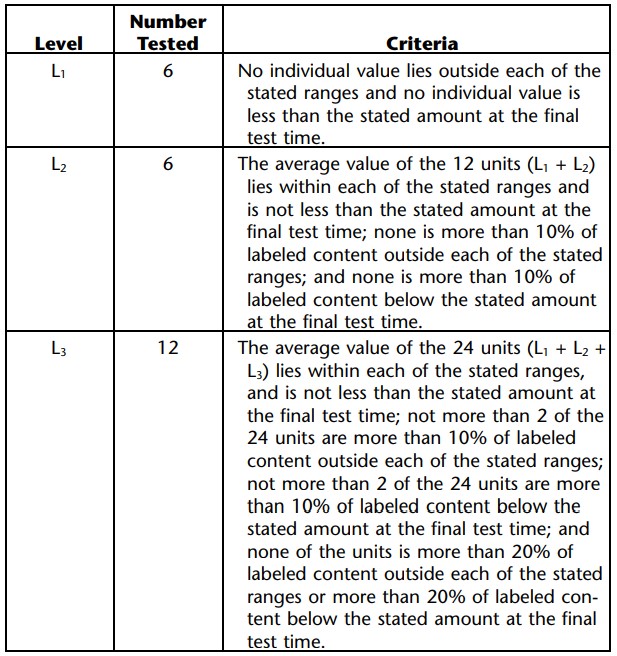
Extended-Release Dosage Forms:
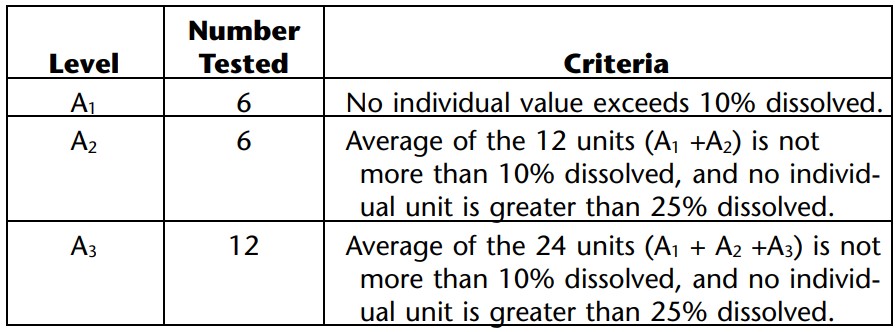
Delayed-Release Dosage Forms:
Q.7 How do you validate an analytical method for HPLC, GC, or other techniques?
Ans: Validating an analytical method requires several ways with particular criteria depending on the method used. Still, using general guidelines helps ensure appropriate validation.
First of all, it is essential to specify exactly the purpose of the analysis and the measuring criteria. For example, using high-performance liquid chromatography (HPLC), let us find the concentration of a specific substance in a sample. Here the concentration of the compound is the parameter of interest since the aim is to quantify it.
Many data points are gathered and examined to verify the HPLC technique. This covers conducting precision studies to assess the repeatability and reproducibility of the method. Multiple injections of a standard solution with known concentrations are done, and the resulting peak regions or heights are recorded. One can evaluate the precision of the procedure by means of statistical computations including the calculation of standard deviation or relative standard deviation.
Accuracy investigations are also carried out to determine the measured values’ relative closeness to the actual values. This may be performed by evaluating samples with known concentrations and comparing the measured results to the predicted values. Many times, statistical calculations including relative error or percent recovery help to measure accuracy.
Method validation also tests other criteria like linearity, limit of detection, and limit of quantitation. Analyzing a series of standard solutions with varying concentrations and obtaining the calibration curve provide the basis for linearity investigations. One evaluates linearity using the correlation coefficient (R^2 value). The limit of detection applies to the lowest concentration of the analyte that can be reliably detected, whereas the limit of quantitation indicates the lowest concentration that can be properly measured.
Q.8 Can you explain the difference between Qualitative and Quantitative analysis?
Ans: Qualitative analysis is the process of identifying the presence or absence of a specific substance in a given sample, on the other hand, the quantitative analysis process helps to measure the amount or concentration of that substance. In other words, we can say, that qualitative analysis answers the question “Is it there?” while quantitative analysis answers “How much is there?” Here, Both types of analysis are used to fully characterize a sample and understand its composition. These both types of analysis are done by using HPLC and GC instruments.
Q.9 How do you troubleshoot issues with peaks on a chromatogram during an HPLC run?
Ans: Peak issues on a chromatogram during an HPLC run may be caused by a variety of factors. For example, if there are air bubbles in the system, they can cause irregularities in the peaks. This issue can be resolved by properly degassing the mobile phase prior to conducting the analysis and verifying that the system is free of any leakage or loose connections.
Incorrect implementation of the column or flow rate settings may be another contributing factor. Wrongly set up columns or either too low or too high flow rate could lead to a distorted peak or broad peak. This can be remedied by carefully installing the column following the manufacturer’s instructions and optimizing its flow rate to match the analyte in question.
Contaminated mobile phase may also cause peak issues. For instance, impurities and mobile phase degradation over time can affect peak shape and resolution. In these cases, a new mobile phase should be prepared and suitably refined.
The injection technique and sample preparation method also have an effect on peak issues. An inadequately prepared sample will cause tailing, while excessive injection volume will lead to split peaks.
In order to prevent this, it is necessary to implement appropriate sample preparation methods and optimize the injection volume in accordance with the analyte’s concentration and character.
Q.10 What are some common sources of error in UV or IR spectroscopy?
Ans: In case of UV or IR spectroscopy in present time, the presence of impurities or foreign matter is a common source of errors. As an example, if there is a little water in the sample there may be some additional peaks that overlap with the desired peaks thus producing inaccurate measurements. One way to eliminate this problem is by properly purifying the sample before analysis, which can be done through techniques such as filtration or solvent extraction for impurity removal.
Another possible type of error is instrumental mistakes which are caused by wrong calibration and defective machines. For instance, if spectrometer is not well calibrated then the reading obtained may be inaccurate. To avoid this problem it is essential to regularly maintain and calibrate spectroscopy instruments. This will involve doing regular checks and adjustments for accurate measurements.
Human mistakes can also result in inaccuracies within UV or IR spectroscopy. This may involve incorrect sampling methods like introducing air bubbles into a sample or just mishandling of the specimen during analysis Incorrect data recording including transcription errors or misinterpretation of findings can also cause impreciseness.
To minimize human error, proper training is essential. This includes providing operators with detailed instructions on proper sampling techniques and ensuring they are familiar with the standard operating procedures. Regular training and refresher courses can also help reinforce good practices.
Q.11 How does sample preparation affect the results of an analytical method?
Ans: The accuracy of the results of analytical methods is impacted by sample preparation. For instance, we will look at pesticide analysis in fruits.
When studying the fruits for presence of pesticide residues, a fruit sample must be selected. As a result, the fruit undergoes processing through washing and peeling to eliminate any surface contamination. The analyte of interest which is the pesticide has then to be extracted from the fruit matrix. This can be achieved using a solvent extraction technique where the pesticide gets dissolved in a suitable solvent after it has been mixed with fruit pulp that contains it. The extract is then made clean by separating any interfering substances that could have contaminated it.
Finally, one may analyze such prepared samples using an appropriate method of detection such as gas chromatography or liquid chromatography coupled with mass spectrometry (GC/MS). Pesticides detected by such instruments are indicated by signals generated as a result of their response.
To interpret this data entails comparing the measured signal with calibration standards obtained from known concentrations of pesticides in order to quantify the analyte present in the sample; this allows us to determine whether or not there are any residues and if so, what their concentration level is like.
Getting proper sample preparation is essential for obtaining accurate and dependable results. If the pesticide isn’t extracted properly or there is any contamination in handling, the concentration measured may be lower than what is actually present in the sample. However, exhaustive sample preparation may better sensitivity, selectivity, and precision to give valid data.
Q.12 Can you give an overview of the USP guidelines for impurity analysis in drug substances or products?
Ans: The United States Pharmacopeia has guidelines for impurity analysis in pharmaceutical products. Specifically, Chapter <1086> of the USP outlines these very guidelines.
An impurity according to USP is any component that is not the intended therapeutic agent or one of its known degradation products. They may come from various sources such as starting material, synthesis process, and storage conditions.
According to these USP guidelines, impurities in drug substances should be identified and quantified in line with GMP (Good Manufacturing Practices) as well as current regulatory requirements. The limit for an impurity in a drug substance is usually less than 0.1% depending on the potential toxicity of the impurity.
However, GMPs also require that for drug products, impurities must be specified and quantified. Any drug product’s specification limit for impurities is typically below 0.05%, again because of possible harmful effects that might result from their presence.
Additionally, it should be noted that USP guidelines have validation requirements for impurity analysis methods. This necessitates using appropriate reference standards and control procedures to ensure accuracy of results.
For more information and specific requirements on drug products’ impurity analysis, refer to Chapter <1086> of USP.
Q.13 Can you discuss the role of quality control in pharmaceutical analysis and how it differs from quality assurance?
Ans: Quality Assurance (QA) and Quality Control (QC) are very important in pharmaceutical analysis. While they are often thought to be the same, there are differences between them.
In other words, quality control is about maintaining desired product quality. This is achieved by analyzing samples at different production levels so as to identify any possible shortcomings or deviations from set specifications. In pharma analysis, QC involves purity, potency, and other critical parameters that ensure that the drug product conforms to requisite standards.
Conversely, quality assurance is a much wider term covering a range of activities geared towards preventing mistakes and ensuring uniformity in quality. These may include creating and implementing standard operating procedures (SOPs), undertaking internal audits, and training staff members.
Q.14 Have you used any software to analyze data from HPLC, GC, or other instruments? How proficient are you in using these tools?
Ans: Software for data analysis has been utilized by me before in my past jobs. Using software like Chromeleon and ChemStation for HPLC analysis and MassHunter for GC-MS analyses are areas of my expertise as well. With these kinds of software, I can easily manipulate and understand huge volumes of analytical instruments’ data. In addition, they possess functionalities such as peak integration, plotting of calibration curves, and exporting data to streamline the process and improve productivity. Moreover, I have some experience in solving problems related to software applications as well as enhancing methods that are useful in analyzing data.
Q.15 Can you discuss a project where you had to troubleshoot issues with equipment during an analytical method development? How did you resolve the issue?
Ans: Yes, during a method development project for impurity analysis in a drug product, we encountered issues with the HPLC instrument. We were not able to get consistent and reproducible results, despite following the same method and parameters.
After troubleshooting, we discovered that the issue was caused by contamination in the HPLC system. To resolve this problem, we thoroughly cleaned and flushed the system with appropriate solvents. We also checked for any leaks or clogs in the system and made sure that all components were functioning properly.
We repeated the analysis using a sample containing known impurities, and this time we obtained accurate and reproducible results. This experience taught me the importance of regularly maintaining and cleaning analytical instruments to prevent contamination and ensure reliable data.
Q.16 Have you ever faced any issues with instrument calibration or maintenance? How did you handle them?
Ans: No doubt, calibration, and servicing of instruments have been my problems. This happened once when we were calibrating our HPLC system only to discover that the standard solution had expired resulting in wrong calibration results.
We had to order a fresh standard solution and repeat the calibration process after receiving it. Whenever we would do any instrument calibrations subsequently, we made a point of ascertaining that all our solutions and reagents’ expiry dates were right at the beginning.
Another example was a blocked GC-MS column which caused difficulties with the instrument. This involved replacement of the column in addition to regular cleaning and changing inlet liners as part of routine maintenance activities.
Q.17 Can you discuss the importance of data integrity in pharmaceutical analysis and how you ensure it in your work?
Ans: Pharmaceutical analysis depends on data integrity so that the results are accurate, dependable, and traceable. Wrong or doctored information could make people to draw false conclusions about the product quality and hence endanger patients’ lives.
I secure my work through Good Laboratory Practices (GLP), which is a set of guidelines enforced in my laboratory. Also, I follow analytical procedures based on standard operating procedures (SOPs). Further, all records inclusive of raw data, calculations, and results are well kept.
Q.18 Have you worked on method transfer for an analytical method? If yes, can you explain the process and any challenges you faced?
Ans: Indeed, I have had some experience in analytical method transfer. In this regard, the establishment of a method should be interchanged from one laboratory or instrument to another without affecting the obtained results.
The process usually commences by grasping the original procedure and its validation parameters. To adapt it to our specific laboratory and equipment settings, we then carry out initial tests on the method. Thus, we may vary such parameters as column temperature, flow rate, or mobile phase composition.
Q.19 Can you discuss the role of method robustness in analytical techniques and its impact on results?
Ans: Yes. In order to ensure method robustness, we use method validation experiments to determine specificity, linearity, accuracy, precision, and stability. We also carry out stress tests aimed at evaluating the impact of extreme conditions on method performance. If necessary, we can adjust the method so as to enhance its robustness.
Q.20 Can you explain the principle behind chromatographic separation in HPLC, GC, or other techniques?
Ans: This chromatographic separation is based on differential partitioning whereby a mixture of compounds separates into individual components by different affinities for two phases i.e. mobile phase and stationary phase. In HPLC, the mobile phase is usually a liquid while in GC both phases are gases with the stationary being solid or bonded material.
Q.21 Can you discuss the role of quality by design (QbD) in method development and validation for analytical techniques?
Ans: QbD is a strategy employing scientific principles for creation and development of analytical procedures. QbD ensures solid and effective approaches by way of understanding critical processing factors as these influence the method’s performance. These experiments optimize methods thereby identifying quality attributes that are important, satisfying all necessary quality standards. Development time and cost have been reduced by QbD but enhanced overall productivity. Furthermore, it assists in resolving difficulties when transferring methods or using them in routine practice. QbD is essential to obtain high-quality and dependable analytical results.
Conclusion:
To sum up, it is very important for you to understand the basic concepts of HPLC, GC, IR, UV, and Mass Spectroscopy if you wish to apply for an analytical chemist Role. Most of the questions from the interviewers are concerning the practical aspects of a role including the use of these instruments, their calibration, procedure, and different troubleshooting procedures (The most important part of your Job). hence, to succeed in interview you must go through their minute details.
Don’t Miss This…
- Top Asked Dissolution Interview Questions with Expert Answers from Pharma Professionals
- D. Pharma Course: Fees, Admission Procedure, College, Scope
- BQS Machine Interview Questions and Answers
- 100+ Quality Assurance Interview Questions Pharma
- Technical Pharmaceutical Interview Questions/Answers
- Top Tips for Face interview successfully
- Top Asked Pharma Packing Questions for the Interview

Naresh Bhakar is the Founder and Author at Pharmaguddu.com, bringing his extensive expertise in the field of pharmaceuticals to readers worldwide. He has experience in Pharma manufacturing and has worked with top Pharmaceuticals. He has rich knowledge and provides valuable insights and data through his articles and content on Pharmaguddu.com. For further inquiries or collaborations, please don’t hesitate to reach out via email at [email protected].