Quality Control Specialist | Industry Experience
✓ Reviewed by: Pankaj Sharma - Quality Control Specialist
Reviewed for Quality Control accuracy, laboratory practices, analytical methods, and technical relevance
📅 Last Updated: January 2, 2023
1.0 Purpose: To lay down the procedure for handling, investigating, and reporting of Out of Specification (OOS) results obtained during laboratory testing. This procedure provides guidance to assure that the investigations are conducted in a timely manner with appropriate depth and are supported by the necessary documentation to perform root cause analysis as appropriate.
2.0 Scope: The procedure is applicable to Raw materials, Packaging materials, In-Process semi-finished materials, Validation samples, Finished products, Stability samples, Qualification on Working standard samples, samples tested as part of Complaint investigations, and Retest materials analysis performed. This SOP applies to OOS results obtained during laboratory testing of the following:
- OOS results obtained for stability samples past their expiration date will be investigated through Phase I to rule out any assignable cause.
- Hold Time Study sample will be investigated through Phase I to rule out any assignable cause.
2.3 This SOP not applicable to the following condition:
- In-process tests that are performed for the purpose of monitoring and adjusting the process (For Example pH, Water/LOD content).
- OOS results observed during the following samples (method validation, method transfer, product development, vendor qualification, swab, organoleptic tests (like odor and taste), and samples analyzed during analyst qualification on the previously approved sample.
- Intended failures to meet established specifications arising from tests performed as outlined in an approved protocol (example: hypothesis testing, failure mode analysis, and testing of innovator sample and etc.).
- For Microbiology-related OOS.
3.0 Responsibility:
- QC officer/Executive- To follow the procedure in an accurate and reproducible manner and check and trend OOS investigation.
- QA officer/Executive- To issue documents
- QC/QA/Production manager- To ensure proper resources are allocated to support the completion of the investigation in a timely manner.
- Department Head- To ensure training and compliance with this Procedure and examination or measurement which fails to meet pre-determined specifications is defined as OOS
4.0 Procedure for investigating Out of Specification (OOS)
4.1 General Points to consider:
- All steps described within the procedure section of this SOP are should be justified, documented, and approved.
- Methodology techniques (such as 5 whys, fishbone, cause/ effect diagrams, etc.) may be employed as necessary in order to facilitate the determination of the root cause.
- All data acquired during an investigation will be retained with the original data record.
- During Phase I, Phase II, and Phase III of any investigation, all actions must be justified based on sound scientific judgment, documented, and approved prior to performing each action. Following any action, conclusions obtained from such actions are documented and approved prior to proceeding with any following action.
- At any stage in the investigation where an assignable cause is not readily, aberrant OOS result is believed to be confirmed, and the potential impact is evaluated. The date of the decision is clearly documented in the investigation.
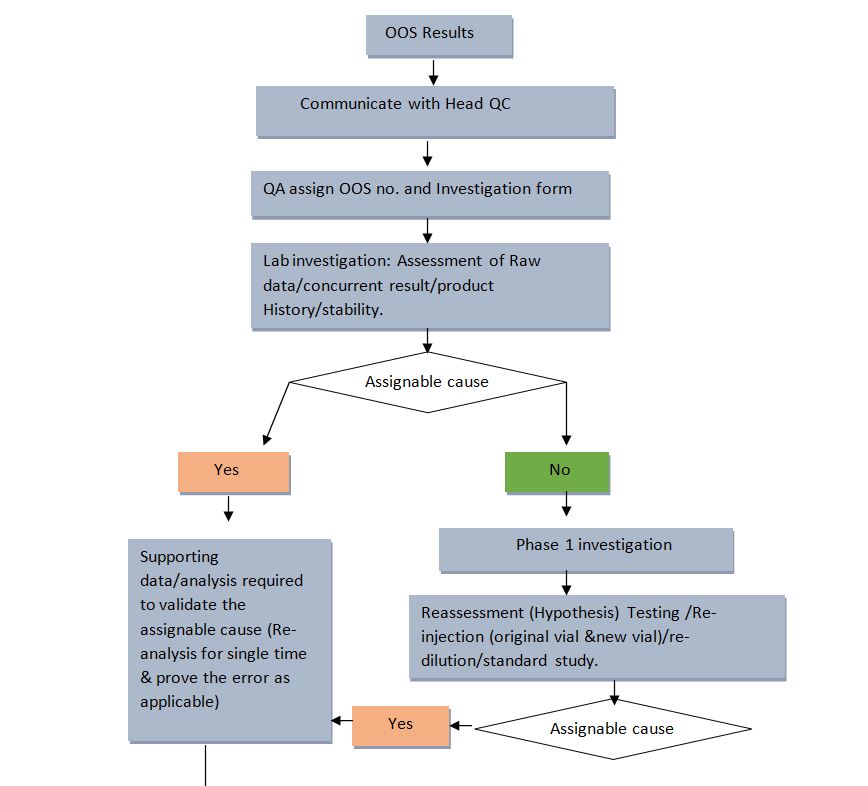
4.2 Initial Identification and Reporting of Out of Specification Results:
- For all analyses, the standards and test preparation must be kept till the analysis result are reviewed by the immediate supervisor / Manager QC. After review, if the result is found to be OOS then the retention of standard and test preparations including all glassware used for analysis to be extended till the completion of the investigation.
- The analyst/supervisor shall report the OOS results immediately to Supervisor or designee.
- The respective Supervisor along with the analyst shall verify preliminary data, recording errors, Calculation errors, and Other data processing errors (such as improper integration of chromatographic peaks) to verify the causes of OOS results.
- If there is an error in the above case analyst shall make appropriate corrections in compliance with established documentation practices.
- The supervisor shall review the corrected result for conformance to established specifications followed by a sign and date on the corrected entries.
- if the verification does not reveal a transcription, calculation, or data processing error, then continue to the next step.
- QC chemist shall notify to QA of the aberrant OOS result and request OOS Investigation forms.
- Site QA shall issue an “OOS investigation report” (Format No. ) to the QC chemist after assigning a unique OOS Investigation Report Number and updating the “OOS tracking log” or (Format No.).
- Out of Specification (OOS) Investigation Report Number shall be assigned as follows:
Z/OOS-XX-YYY
Where:
Z- Manufacturing Site
OOS Out of specification
XX – Indicates the last two digits of the current year e.g. 22 for 2023
YYY – Serial number of OOS in a year, issued as per OOS log (001,002,003….).
Related: SOP on operation and calibration of autoclave
- The supervisor immediately shall interview the analyst, regarding the execution of the analytical procedure, instrument handling, and calculations are done based on the checklist provided in the OOS investigation report.
- The Analyst interview is to be conducted by carefully reviewing the applicable test procedure with the analyst and confirming that the analyst followed the procedure as per the given SOP.
- The QC Supervisor shall perform a historical data review and examine such as all stability data of lots., release date of other similar product lots, working standard area against comparison studies for previous analysis, and historically related investigation reports that involve the same product, same laboratory equipment, same test method, same analyst, etc.
- QC supervisor shall review all concurrent tests performed for the investigated material (e.g. if the result being investigated was for assay testing, the results for other tests such as impurities, pH, moisture, dissolution, content uniformity, etc. should be thoroughly reviewed for conformance to specifications).
- If an assignable cause (Obvious error) is found, Supporting data/ analysis is required to validate the assignable cause. Re-measure, Re-scan, Re-read, and Re-inject the same standard and sample solutions to prove the error (Repeat the same error). If the root cause is confirmed, Re-analyze a single time from the same original sample as per SAP). If required, suggest CAPA.
- If an assignable cause is not identified during the assessment of Raw Data, Concurrent Results, Product History, and Stability then proceed for Phase I investigation.
4.3 Phase I Investigation:
- QC Supervisor authorizes the steps to be performed for Phase I. Phase I testing plan is not limited to hypothesis analysis like re-injection/rescan, re-fill vial, and re-dilution.
- Failure mode analysis if an assignable cause is not identified during the initial hypothesis. then plan for additional testing as justified. Re-mix and Re-Measurement, Re-mix, and Re-dilution (if applicable). study and compare the initial standard response, area, peak pattern, and height.
- Analyst-Related KLE (Known laboratory error) shall be checked.
- Analyst-Related Known laboratory error shall be one of the following but not limited to:
- The incorrect procedure followed during solution preparation or non-compliance to critical steps during analysis.
- Improper precautions followed during the analysis
- Any specific techniques included in the analytical procedure which has not been used by the analyst
- Improper sample handling/Storage handling during analysis
- Improper sonication or extraction during sample preparation
- This “Phase I Investigational Testing Plan” is to be documented and approved by the QC Supervisor and QC Manager.
- The Analyst shall re-analyze the original sample preparation as described in the Phase I Investigational Testing Plan. All Phase I testing is documented in the laboratory worksheet and related records in accordance with respective site-specific SOP for Good Documentation Practices, including a reference to the investigation number.
- Upon completion of the Phase I Investigational Testing Plan, the QC Supervisor and the Analyst jointly review the results and determine if an assignable cause has been identified. The QC Supervisor records the results of the Phase I Investigational Testing Plan promptly on Format indicating that the results confirm that the result is attributable to the assignable cause. When the result is attributable to an assignable cause, the cause is corrected, and the test is repeated two times on the original sample and proves the error as applicable. Upon satisfactory completion of the repeated test, the repeated test result is reported on the COA. An explanation will be documented in the laboratory worksheet and both the analyst and QC Supervisor will initial and date the worksheet entry. The initial testing results are invalidated.
- The QC Supervisor records the impact of the Phase I Investigation on the associated impacted batch or batches. Phase-I Investigation results clearly show an instrumental error or a standard solution error as the assignable cause will require the impacted batch (es) to be invalidated and retested. Phase 1 Investigation results that show the OOS result is only present in the test batch support the release of the associated batch (es).
- Upon completion of the initial data assessment, if the Out of Specification result involves the testing of a marketed product as an ongoing stability study, a Notification of Potential Product Failure shall be initiated by QC Manager/designee by using Format.
- Notification of potential Product Failure:
- Site QA shall send the notification of OOS.
Note: Upon completion of Phase I Investigation if an assignable cause is not identified we go to phase II:
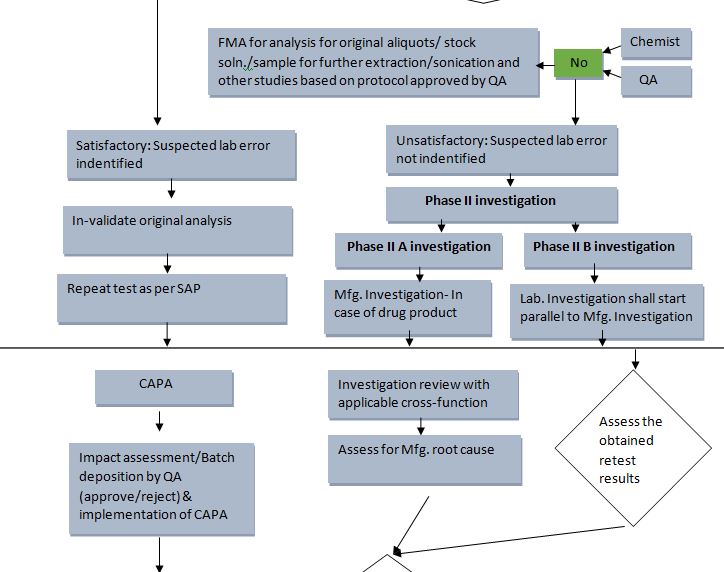
4.4 Phase II Investigation:
It contains two-part
- Phase II A: Manufacturing investigation
- Phase II B: Laboratory Investigation testing plan for re-test.
4.4.0 Phase II A: Manufacturing investigation
- The full-scale Manufacturing Investigation team shall review the Batch record, History Data, and document.
- The designee QA/QC, manufacturing, engineering, process development, cross-functional SME, etc. shall conduct a shop floor investigation which includes a review of the process and procedure.
- If the assignable root cause is found during the investigation then the investigation is concluded followed by initiation of deviation and further impact assessment/ Batch Disposition by the Site Quality Head (Approve/ Reject) & implementation of CAPA, however, the investigation must be extended to other batches of the same product or other product that may have been associated with the specific failure shall be investigated.
- Laboratory investigation shall start parallel to the manufacturing investigation.
4.4.1 Phase 11 B Laboratory Investigation:
- No obvious laboratory error exists and no extraction, dilution, injection, or standard preparation errors are evident as determined in Phase I. Thus, the investigator will initiate a retest with the goal of determining the assignable cause of the OOS result(s). This review shall be documented.
- Following the retest, if the initial result(s) is confirmed further testing must be justified based on the probable assignable cause. All actions must be justified based on sound scientific judgment.
- The typical path for an investigation should proceed as described below. Based on sound scientific judgment, other investigation tests may be performed for the purpose of discriminating between sample preparation, instrumental, and methodology errors. Each step within Phase II must be designed, justified, and approved prior to proceeding with the analysis. It should be noted that repetitive Phase II test plans are not permitted without a scientifically justified assignable cause theory. Approval must be obtained from a QC manager or designate in consultation with a site quality head or designee for all portions of Phase II.
- If sample preparation errors are suspected, the sample may be retested from the original laboratory composite. The number of sample solutions prepared for any Re-test should be justified and sufficient to confirm or refute the aberrant result(s). Unless otherwise documented in the investigation, the number of samples prepared for the Re-test will be six (6) replicates.
- The retest may be performed by the Second qualified analyst (preferably in the presence of the First analyst). The identity of the analyst responsible for the retest will be clearly documented. Participating in the investigational testing will prepare individual independent sample preparations for each retest. The analyst will establish all System Suitability and Continuing Calibration criteria independently for their testing, in accordance with the standard analytical procedures (SAP).
- Proceed with Confirmation Study testing (Re-test) with n=6 preparations of the original sample. Note: If the original sample has been consumed, an appropriately acquired alternate sample may be utilized in consultation and required approval from Site Quality Head or designee.
- The analyst will independently prepare any solutions relevant to the investigation, such as their own Diluent, Stock Solution, Mobile Phase, and Sample preparations required by the method, and will perform the analysis in accordance with the SAP.
- If an instrumental variation is relevant to the design of the investigation, the analyst will use different instrumentation, wherever possible. Preparation of independent Standard preparations may be required when the Standard preparation is relevant to the design of the investigation.
- If a methodology/technique error during retest is noticed, the same shall be documented and referred to the Analytical development team & CAPA to be initiated.
- The QC Manager reviews the results of the six replicates and records the results.
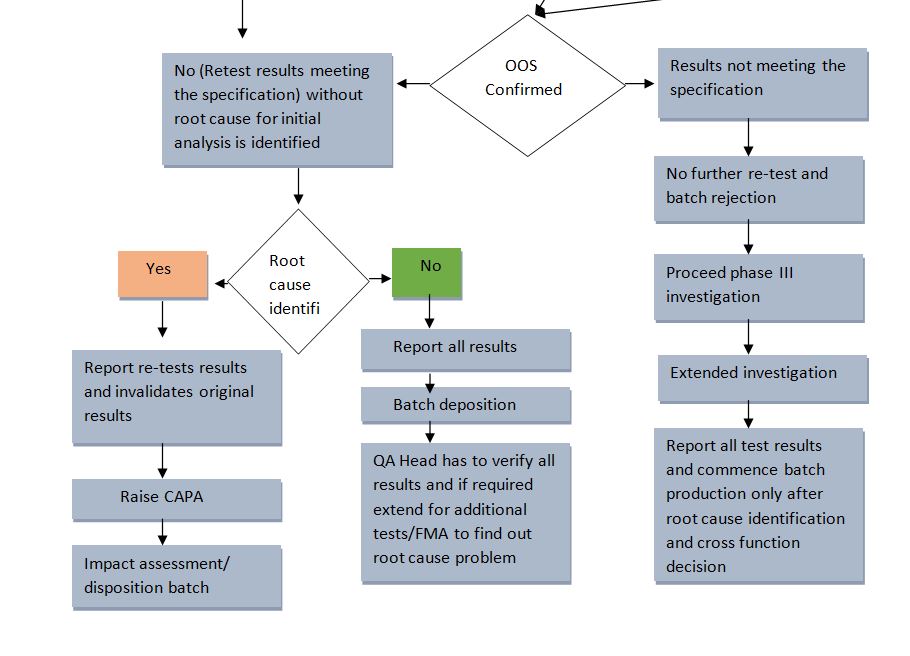
- In case the re-testing confirms the OOS then, no further testing is required and results can be reported.
- In case of OOS doesn’t conform but an assignable cause is found then, the QC analyst shall invalidate the original results after approval by Site Quality Head. This final result (Average of 6 replicates) shall be reported and appropriate CAPA shall be raised for rectification of the root cause.
- In case of OOS doesn’t conform and an assignable cause is also not found then, all results of initial analysis and hypothesis testing shall be reported. The site Quality Head has to revisit all results before taking a decision for batch disposition. Wherever necessary protocol-based additional testing plan and failure mode analysis are to be initiated from the original or control sample with strong technical/ scientific knowledge to justify batch disposition by the Site Quality Head/ Designee.
- If the assignable cause has not been identified during the shop floor investigation and laboratory investigation, then the next course of action (Extended investigation) would be decided by the Site Quality Head/ Designee. If the assignable cause has not been identified, during in-process testing of the drug product batch can be monitored (based on deviation) for the next manufacturing stages i.e. compression/coating / FO for critical quality attributes of the product based on stratified analysis testing plan may support to identify the product homogeneity and outliers. In the case of drug substance, the vendor’s results and history of the vendor to be reviewed for any similar occurrence, and based on the evaluation batch disposition decision shall be decided.
- In the case of RM/ PM. Out of Specification is confirmed (valid) at Phase I and II hence no further retest is required. In case a joint analysis (mutually agreed by Company and Vendor) is required then it shall be approved by the Site Quality Head/ Designee. Otherwise, the batch disposition decision shall not require Phase III Investigation.
4.5 Extension Request and Interim Status Summary:
- The entire laboratory investigation should be completed within 30 business days. if the investigation to required more days, the department needs to request along with formate no.
- The approval of the Head CQA or designee is required for any extension beyond the second extension.
4.6 Phase III Investigation (Sampling Error Investigation) for Drug Products only:
- In Phase Ill investigation, the cross-functional team shall evaluate Manufacturing Phase IIA and Laboratory investigation Phase 11B.
- Sampling error Investigation (Only If deemed necessary by the outcome (s) of the Phase!! investigation) may be performed to determine the root cause.
- The site Quality Head or designee shall take the decision for determining the sampling error
- If the Sampling error is established (Only if deemed necessary by the outcome(s) of the sampling error investigation) Re-sampling plan shall be prepared. Site Quality shall raise a request for re-sampling.
- Re-sampling is not considered an automatic next step of the investigational process even when the assignable or root cause is not found as part of the laboratory investigation.
- Re-sampling only be considered if it has been determined at the point of investigation reveals the original sample was improperly sampled, and stored (if not, it may be affected by exposure to humidity. light, or heat).
- Based upon the intimation gathered in the review of the OOS investigation and relevant scientific principles, then the QC Manager will develop an Additional Testing Plan to evaluate potential root causes for the OOS result attributable to laboratory sources. The Additional Testing Plan/ Failure Mode Effective Analysis based on the protocol is to be performed on the original laboratory sample if sufficient material is available. This Additional Testing Plan is to be documented.
Note: The stability of the standard and sample solutions should be considered (If available based on the reference of Validation Report or Pharmacopoeia If any) and documented when performing investigational testing. (This is extremely important to consider for sensitive testing such as impurities or related substances). - Obtain QA approval of the Additional Testing Plan or an alternate protocol providing a description of the testing, investigation requirements, samples to be tested, and how the data will be evaluated. Modifications to the procedure may be necessary to determine the root cause.
Note: The results of the Additional testing may not be used to replace an original suspect analytical result. It may only be used to confirm or discount a probable root cause to identify aberrant OOS (for information only). Determine if the assignable cause of the aberrant OOS Result has been established. If a root cause has been identified, propose corrective action. - Evaluate the results of the Failure Mode /Additional Testing Plan. If the retest complies with the specification, Proceed, Closure of Laboratory Investigations. Commence batch production only after root cause identification and cross-functional decision followed by OOS Investigation report, CAPA, impact assessment, and batch disposition.
4.7 Laboratory Operations / Quality Impact Assessment:
- Determine the impact or scope of the laboratory investigation by evaluating the following items to identify another batch(s) or material(s) potentially implicated by the investigation.
- were the material(s) tested using the same laboratory equipment?
- were the sample(s) tested using the same reference standards or reagents?
- Was the sample(s) tested by the same personnel?
Note: The items listed above are not all-inclusive and are only used as a guide. Determine if any additional factors are relevant to a complete impact assessment, as needed.
- If the conclusion is made that a system deviation has been identified, then a Quality Impact Assessment is conducted.
- The Quality Impact Assessment will include the following information:
- All potential adverse impact related to analytical data, Good Manufacturing Practice (GMP) compliance, and sample integrity is reviewed.
- The appropriate time period affected by the adverse event is identified.
- A review of all investigations is conducted to determine if any relationship is present between these issues and the system deviation.
- A review of relevant SOPs is performed and any necessary revisions will be identified and conducted in a timely manner.
- A review of current laboratory practices and policies will be reviewed in order to identify the need for remediation of current procedures or the creation of new procedures.
- A review of any validation and supporting documentation is performed in order to identify needs for revision. Changes are submitted and approved in a timely manner.
- A review of any methods and specifications will be performed in order to identify the need for revision. All relevant change control will be conducted in a timely manner.
- Any necessary training or re-training will be identified and conducted with the appropriate personnel.
Related: Change control SOP
- A comprehensive historical data review is performed in order to evaluate any potential effect on data due to the system deviation. Attachments including product release history, stability data, and recalculated results for comparison are included with this portion of the evaluation.
- Any consultation or contract service from an outside party may be conducted, if applicable. Any work performed in this capacity will be justified and thoroughly documented.
- Based on the results of the previous steps, a decision will be made as to the potential adverse Quality Impact. If no Quality Impact has been determined, the inquiry is closed and such findings will be documented in the conclusion of the Quality Impact Assessment. If an adverse Quality Impact has been determined, the affected data is identified within the conclusion of the Quality Impact Assessment and the Quality Impact Assessment will require approval from the Site Quality lead or designee.
- Corrective Action / Preventative Action (CAPA): The investigator and supervisor should identify any errors due to the analyst’s training, technique, practices, instrument performance, or test methodology. To prevent reoccurrence, appropriate corrective actions / preventative actions should be initiated by creating a CAPA, tracking, and trending.
- Document impact assessment and applicable actions in the Laboratory Investigation Report.
4.8 Batch Disposition:
- The final batch disposition decision should be made by the Site Quality Head/ Designee within 30 working days from the discovered date of the Out of Specification. The following parameter shall be investigated prior to batch disposition:
- Investigation conclusion
- Root cause summary
- The decision for batch disposition determined
- A review of events during the production of the batch reveals no aberrations or indications of unusual process variation
- Extended investigation including re-sampling and additional testing can be planned as per approved protocol by the Site Quality Head (If required) for information only.
- All results shall be reported. Based on the investigation outcome, the probable root cause(s) identified/ CAPA(s) with proper technical/ scientific and strong justification provided by CFT, and the decision for approval (Further batches shall be commenced for production).
- The batch disposition decision by Site QA shall be based on the outcome of Phase I, II, and III investigations. Note: Average results are to be reported as per pharmacopeia! criteria for stage testing. For other tests, if obtained any of the test results as outliers, all repeat test results are to be average or report all test results, wherever applicable.
- Depending upon the assignable root cause, corrective action or actions may be developed to correct the issue identified and to prevent similar future recurrences.
- Whenever applicable, open a CAPA as per CAPA SOP. document the CAPA number on the Out of Specification report.
4.8 Closure of Laboratory Investigations:
- The QC Manager completes OOS Investigation Report.
- Include any Extension Requests and Interim Status Reports prepared.
- Include an Impact Assessment, Laboratory Operations Impact Assessment —OOS Results, providing attachments as needed to document fully the impact assessment.
- A repeated assignable cause identified shall be re-classified for a more specific reason based on trend analysis from the last two years to assess the OOS and linkage for repeat events and CAPA effectiveness.
- Include any resulting CAPA developed, Laboratory Operations Impact Assessment OOS Results.
- Indicate the QC compliance team (QCMT) status of the batch.
- Review and approve the completed Format for Deposition.
- The Site Quality Head reviews and approves Investigation Summary and Product Disposition Format during this review.
- On batch certificate reference of investigation shall be given i.e. OOS investigation number shall be provided on batch CoA for batch record review.
4.9 Post-Approval Changes:
Addendum: An addendum may be added to the OOS record to capture additional comments or ancillary documentation pertinent to the record. If the disposition of the material or the root cause conclusion is to be changed, the record must be re-opened and approved by the site quality head or designee.
- The QA Management or designee updates the appropriate OOS Tracking Log in (Out of Specification tracking log) with the completion date.
- Upon completion of the final data assessment, if the confirmed OOS result involves the testing of a marketed product either in an ongoing stability study, Notification of Product Failure shall be initiated by QC Manager/designee by using Format, Notification of Product Failure on confirmed Out of Specification (OOS).
- For OOS sequence in LIMS shall be recorded as per site-specific SOP for sample workflow in LIMS.
4.10 OOS Investigation procedure flow for Blend Uniformity Testing:
- The analyst shall notify the respective Laboratory Supervisor of an aberrant OOS obtained from blend uniformity testing in Stage 1. A Stage 1 blend is OOS i.e. results obtained in the blend uniformity test are not within the established specification range.
- The analyst shall participate in Phase I Investigation with the assistance of the QC supervisor. Determine which of the ten units contributed to the aberrant OOS of the blend uniformity.
- In case the OOS result is obtained in any unit (N=10) sample, investigational analysis of failed unit shall be done to identify any KLE.
- During investigational analysis, if KLE is identified and the unit passes, OOS confirms to be invalid then appropriate corrective action is to be implemented.
- The second set of ten locations is tested and the blend is to be released upon confirmation of passing laboratory results.
- If repeat testing does not conform with established acceptance criteria, then an examination of the results is to be done to determine which of contributing unit (s) tested contributed to aberrant OOS of blend uniformity. The contributing unit (s) will be examined through phase I Investigation to eliminate any KLE and confirm the OOS.
- In case failed unit fails during investigational analysis and confirms that there is no laboratory error with initial results obtained, but the results obtained comply with stage 1 acceptance criteria (i.e. Mean of the blend is within 90.0 % to 110.0 % of the expected quantity of active ingredient with a relative standard deviation (RSD) of not more than 5.0 percent), initial results to be considered for reporting results and proceed to phase II manufacturing investigation and parallel to it start stage 2 testing for a total of twenty (20) additional samples.
- If no assignable cause is identified in Phase II manufacturing investigation and stage 2 (20 additional samples for blend uniformity test) i.e. if all 20 individual sample resting results, stage 2 acceptance criteria (mean of the blend is within 90.00% to 110.0%) and percentage RSD is complying as per criteria then based on obtained results and QA evaluation, a deviation can be logged. On approval of deviation, the batch can be further processed.
- Stage 2 testing requires the testing of 2 more dosage units per location for a total of twenty (20) additional samples. The twenty samples are tested per the appropriate SAP.
- The average and RSD of both Stage 1 and 2 (n=30) is determined. The blend conforms if the mean of the blend is within 90.0% to 110.0 % of the expected quantity of active ingredient to RSD is <5.0%.
- If the blend meets the acceptance criteria for the Marginal criteria method (MCM) the blend is recommended for release. If the aberrant OOS is confirmed and the acceptance criteria for MCM are not achieved the blend is recommended for batch disposition by Site Quality Head.
4.11 OOS Investigation procedure flow for Dissolution Testing:
- Follow the site-specific dissolution start-up checklist before preparing dissolution media.
- Log any error or abnormality during media preparation through laboratory incident SOP for “Handling of Laboratory Incidents“.
- Notify a laboratory supervisor of an aberrant OOS test result obtained from dissolution testing.
- If results are not meeting the specification at SI/ LI/ AI, B1 stage, review the data based on the below criteria (but not limited to), refer to 5.40.12:
- If any significant change, 1 or more dissolution units from a total of 6 units found on the lower or higher side of the specification limits
- Batch results or trends not meeting as per historical batches.
| Stage (for Immediate-Dosage forms) | No. of Dosage tested | Accpt. Criteria |
|---|---|---|
| S1 | 6 | Each unit is not less than Q+5% |
| S2 | 6 | (S1+S2) Avg. value of 12 units is equal to or greater than Q, and no unit is less than Q-15 %. |
| S3 | 12 | (S1+S2+S3) Avg. value of 24 units equal to or greater than Q, and no more than 2 units is less than Q-15 % and no unit is less than Q- 25%. |
| Stage (for Extended-release Dosage forms) | No. of Dosage tested | Accpt. Criteria |
|---|---|---|
| L1 | 6 | No individual value lies outside each of the stated ranges and no individual value is less than the stated amount at the final test time. |
| L2 | 6 | (L1+L2) Avg. value lies within each of the stated ranges and is not less than the stated amount at the final test time non is more than 10% of labeled content outside each of the stated ranges, and non is more than 10% of the labeled content below the stated amount at the final test time. |
| L3 | 12 | (LI + L2 + L3) The average value of the 24 units lies within each of the stated ranges and is not less than the stated amount at the final test time; not more than 2 of the 24 units are more than 10% of labeled content outside each of the stated ranges; not more than 2 of the 24 units are more than 10% of labeled content below the stated amount at the final test time, and none of the units is more than 20% of labeled content outside each of the stated ranges or more than 20% of labeled content below the stated amount at the final test time. |
| Stage (for Delayed-release Dosage forms) | No. of Dosage tested | Accpt. Criteria |
|---|---|---|
| B1 (Acid Stage) | 6 | No individual value exceeds 10% dissolved |
| B2 (Buffer stage) | 6 | Each unit is not less than Q+5% |
- If S1/L1 stage test results fail in the above-mentioned criteria, the analyst/ reviewer shall calculate the results based on specification time points/ limits considering the values for S2/ L2 stage as per general pharmacopoeia dissolution criteria.
- If calculated results meet the general pharmacopoeia dissolution criteria for the stage (level 2), shall proceed for further stage (S2/L2) testing without any further investigation. Note: No OOS/ No Incident is required in this case, to repeat S2/ L2, analytical worksheets are to be part of the request followed by approval from the Head site QC.
- If all dissolution units (6+6=12 units) test results and average result complies with general pharmacopoeia dissolution criteria, report as Dissolution complies at S2/ L2 stage.
- If S1/L1 & S2/L2 stage testing results didn’t meet the specification limits, the analyst/ reviewer shall calculate the results based on specification time points/ limits considering the values for S3/L3 stage as per general pharmacopeia dissolution criteria.
- If calculated results meet the general pharmacopoeia dissolution criteria for the stage (level 3), shall proceed for further stage S2/L2 (6 units if not tested) and S3/L3 (12 units), a total of additional 18 units testing without any further investigation.
Note: No OOS/ No incident in these cases, to conduct S2/L2 & S3/L3, analytical worksheets are to be part of the request followed by approval from the Head site QC and Head Site QA.
- If all dissolution units (6+6+12=24 units) test results and average result complies with general pharmacopoeia dissolution criteria report as Dissolution complies at S3/L3 stage.
- Level 3, batch disposition shall be based on product historical data / CAPA/ stability with scientific justification by Head Site QA.
- In case, S1/L1 stage results fall in the above-mentioned criteria in point 5.40.4, and calculated results based on specification time points/ limits considering the values for S2/L2 & S3/L3 stage testing as per general pharmacopeia dissolution criteria did not meet the general pharmacopeia dissolution criteria, OOS shall be logged and investigated as per the procedure.
- Hypothesis analysis mandated to be checked as part of Lab Investigation:
- OOS certified investigator (role specific), to check the bench top solutions as per test method for physical verification and plan to OOS Phase-1 flow chart
- Re-inject the same HPLC vial
- Re-fill the same (final solution) into the HPLC new vial and inject
- Re-dilute from the original stock solution to further dilution & re-fill into the HPLC new vial and inject (if the original stock solution availability)
- Same standard solutions to be injected (standard evaluation)
- Standard & sample solution stability check
- Historical trends (if any) for regular / stability batch analysis Note: Based on hypothesis analysis, KLE (if any) identified as a root cause (ex: wrong dilution, instrument error, etc.,), same shall be re-confirmed based on similar/ simulation study as “failure mode analysis”, extended the phase-I investigation to prove/ confirm KLE as identified reason with suitable justification.
- If identified KLE reason is justified, initial test results shall be invalidated and the same batch shall be retested for a new set of SI/LI stage analyses.
- If test results are found to meet the specification limits, the batch shall be released based on the final investigation outcome and concluded as the batch complies at SI/LI stage shall conclude initial SI/L1 to be termed as – Invalid OOS. Note: 1. Based on hypothesis analysis and failure mode analysis, if no obvious KLE reason is identified as a root cause, shall test the additional 18 units (S2/L2— 6 units & S3/L3 — 12 units). 2. ‘The acceptance criteria shall be followed as per the respective pharmacopoeia (S1/ LI/ AI, B1, and next stages as per dissolution general chapter).
- To conduct S2/L2 & S3/L3, analytical worksheets are to be part of the request followed by approval from IIead site QC and Head Site QA.
- If the dissolution meets the acceptance criteria for Level 3, then report the 24 units’ results.
- The outcome of the test results: Report all the test results (24 units from all stages SI/LI (6 units) + S2/L2 (6 units) + S3/L3 (12 units)), conclude as an outlier case, and charge for stability (if in case 1st time) to monitor further stations.
Note: Batch shall not be dispatched to any market, without scientific justification by Head Site QA, Head QCMT, Plant Head, and Head CQA.
- If the OOS is confirmed and the acceptance criteria for Level 3 are not achieved, then the dissolution test results are confirmed as valid Out of Specification.
Note: Test sample solution from the initial (SI/ L1/ AI, B1 stage testing) to be retained for investigation until acceptance criteria are met.
- Any ongoing manufacturing/ testing/ batch analysis to be reviewed specific to product/ strength arc part of risk assessment and conclusion for batch disposition shall be under the Site Quality Head.
- The investigation shall be extended to the manufacturing end as per OOS flow chart Phase-11A, as part of an investigation, vendor (API) source evaluation and process changes, and other chances of probabilities (if any).
- Unless CAPA and process controls are not justified, no further batches shall be manufactured at the site for specific products/strengths.
- At any stage, if the confirmed OOS fails to meet the acceptance criteria, the ongoing subsequent stages of the manufacturing process or testing (core/ coated) are suspended, and the batch disposition by the Site Quality Head.
- The batch shall be rejected based on the final investigation outcome — Valid OOS.
4.12 Out of Specification (OOS) Investigation procedure flow for Content Uniformity Testing;
- Notify a Laboratory Supervisor of an aberrant OOS obtained from content uniformity testing.
- Participate in Phase I Investigation with the assistance of the QC supervisor. Determine which of the 10 units tested (LI) contributed to the aberrant OOS of the Content Uniformity test.
Note: The acceptance criteria shall be followed as per the respective pharmacopoeia (AV value: LI/ L2 as per content uniformity general chapter).
- If the aberrant OOS is demonstrated to be a KLE, appropriate corrective action is implemented.
- The second set of 10 units (1,1) is tested and the content uniformity is released upon confirmation of passing laboratory results.
- If the Phase I Investigation confirms the aberrant OOS, an additional set of 20 units (L2) are tested through Level 2 testing (L2) as per SAP.
- The acceptance criteria shall be followed as per respective pharmacopoeia.
- If the content uniformity meets the acceptance criteria for Level 2, the content uniformity is released.
- If the OOS is confirmed and the acceptance criteria for Level 2 are not achieved, the batch is rejected.
- At any stage, if the confirmed OOS fails to meet the acceptance criteria of subsequent stages, the testing of subsequent stages is suspended, and the batch disposition by Site Quality Head.
4.13 OOS Investigation procedure flow for criteria base Physical Testing:
- Notify a Laboratory Supervisor of an aberrant OOS obtained from physical testing.
- Participate in Phase I Investigation with the assistance of the QC supervisor. Determine which of the units tested contributed to the aberrant OOS of the physical testing.
- If the aberrant OOS is demonstrated to be a KLE, appropriate corrective action is implemented.
- A 2nd physical test is tested and the batch is released upon confirmation of passing laboratory results.
- If the Phase I Investigation confirms the aberrant OOS, the physical test is repeated as described in the appropriate compendia monograph.
- The acceptance criteria are per the current USP or EP monograph.
- If the extended physical test meets the acceptance criteria for expanded testing the batch is released.
- If the results of extended testing do not conform to the established acceptance criteria, an examination of the results is done to determine which of the unit(s) tested contributed to the aberrant OOS of the physical testing. The contributing unit(s) will be examined through Phase I Investigation to eliminate any KLE and confirm the OOS.
- If the OOS is confirmed and the acceptance criteria for an extended physical test are not achieved, the batch is rejected.
- At any stage, if the confirmed OOS fails to meet the acceptance criteria of subsequent stages, the testing of subsequent stages is suspended, and the lot/batch disposition by Site Quality Head.
4.14 Trending of Out of Specification (OOS) Investigation:
- Trending of OOS results shall be done periodically category wise like analyst error, instrument error, method/specification error, material/product, etc. The review on trending shall be done quarterly and reported annually as part of the Annual Product Quality Review.
- Sorted by the analyst, shall be reviewed by members of the Quality Management in order to identify any trends in analyst performance. Repetitive occurrences of three or more related events in a quarter will constitute a potential Trend. Upon recognition, a collective decision regarding appropriate corrective/ preventive action will be made in accordance with the guidance on analyst performance evaluation. The report will be provided to the site QC Head, Training personnel, and Site Quality head.
- For analysts who exhibit extensive technical difficulties, the following guidelines will be established based on the detail within the quarterly reports :
- Analysts having three or more related events will be re-qualified on the specific practice or equipment.
- If a repeating trend is identified for an analyst in two consecutive quarters, the analyst’s job responsibilities and qualifications will be re-evaluated.
- Sorted by product and test. The report will be reviewed by members of Quality Management in order to identify any trends in method or product performance. Repetitive occurrences of three or more related events in a quarter will constitute a potential trend. The report will be provided to personnel within Regulatory Affairs and the method validation department (ARL).
- Sorted by instrument. The report will be reviewed by members of the Quality team in order to identify any trends in instrument performance. Repetitive occurrences of three or more related events in a quarter will constitute a potential trend. The report will be provided to the concerned person and in turn, will be discussed with the respective service engineer.
- During the trending, a review of the earlier batches of the same stage shall be carried out to find the effect of the various factors resulting in OOS results.
- In a quarterly report, preventable assignable causes which has three or more events will be re-classified into more specific causes to be able to define the associated actions with even greater detail.
- All OOS with open investigation shall be monitored and followed up by the site quality department.
- Review the laboratory investigations for trending on a quarterly basis.
Annexure 1 for OOS investigation Work Flow:
5.0 Abbreviations:
QC- Quality control
SAP- Standard analytical procedure
No.-Number
CAPA- Corrective and preventive action
Mfg.- Manufacturing
KLE- Known laboratory errors
OOS- Out of specifications

Pankaj Sharma — Quality Control Specialist
Pankaj Sharma is a pharmaceutical QC professional who reviews our technical content for GMP accuracy, laboratory practices, and regulatory relevance.