GMP & Pharmaceutical Manufacturing Expert
✓ Reviewed by: Satpal Singh - Quality Assurance & Regulatory Specialist
Reviewed for GMP compliance, quality systems, and regulatory accuracy
📅 Last Updated: June 9, 2026
ALCOA stands for Attributable, Legible, Contemporaneous, Original, and Accurate – these were the first five principles of the GMP record keeping. For electronic data management, there is an additional “ALCOA+” introduced for Complete, Consistent, Enduring, and Available. “ALCOA++” additionally incorporates Integrity, Robustness, Transparency, Accountability, and Reliability – addressing AI, cloud systems, and advanced digital settings. All three frameworks are expected by FDA, EMA, MHRA, WHO, and PIC/S during GMP inspections.
ALCOA, ALCOA+, and ALCOA++ are related to data, either on paper or electronic form, and defined by FDA guidance.
Key Takeaways:
- ALCOA was introduced by FDA in the 1990s. Next came ALCOA+ with the advent of digital systems in GMP environments. ALCOA++ is relevant to today’s manufacturing in the AI and cloud era.
- On 7 July 2025, the European Commission published a draft revision of Annex 11 and a revised Chapter 4 on documentation and records together with the brand new Annex 22 on AI-based systems – the most significant modernization of the EU GMP digital compliance framework in 14 years.
- EU GMP Chapter 4 2025 documents ALCOA++ principles as key to the proper management and control of documents and records – including paper, electronic, and hybrid.
- Almost 70% of all the critical findings in today’s EU GMP inspections are now data integrity and computerized system control gaps.
- In the new EU GMP Annex 22, the first EU regulatory document for AI systems to be used in pharmaceutical manufacturing, it is again emphasised that the ALCOA++ principles should be applied in all processes involving the use of AI. Qualitest Group
- The revised EU GMP Annex 11 is likely to be published in the middle of 2026 and finalized.
What is Data integrity in Pharmaceuticals?
Data integrity is the key element in pharmaceuticals to ensure products meet all the quality standard parameters throughout the end of products. It is the process of maintaining and assuring the promise and consistency of data over its whole life cycle.
What is the common Data integrity issue?
Two types of common Data integrity issues are as follows:
- Intentional
- Non-intentional
- Intentional issue: To ensure that the regulatory standard is maintained, faculty should have cGMP and a defined procedure. The one that is working in an organization who intentionally does not follow the cGMP and the defined procedure knows that he/she is committing a crime. In such cases, facility/organization to terminate the person who’s doing wrong with data (such as form 483 and a warning letter impacts). A failure to adhere to cGMP and regulatory compliance by intentional data integrity problems is the opposite of what is desired. In the pharmaceutical field manipulation, alteration, falsification, deletion, and modification of the data to achieve the desired final result at acceptable level is not allowed.
- Non-intentional issue: The non-intentional issue may occur due to lack of knowledge, training and experience. In such cases, the organization can perform a root cause analysis on Data integrity issues. If it is found guilty then the organization will have to provide the proper training to the person to avoid Data integrity issues.
Why is Data integrity important at that moment?
Wherever Data integrity issues are discovered, the regulatory bodies take action due to violations. Action may be includes warning letters and holding of imports. The recently large number of warning letters issued by regulatory is related to Data integrity issues.
In March 2015, the medicinal health product regulatory agency MHRA released a new guide on “GMP Data integrity definition and guidance for industry” these regulatory set deadlines to comply with the Data integrity issue up to the end of 2017.
Well, there has been some guidance on good manufacturing practices (21CFR 210, 211 and 212), but FDA published their guidance on Data integrity and compliance with cGMP guidance for industry later in April 2016. Data integrity issues were also referred, but due to lots of regulatory issues and every new update more focus on it.
What Is ALCOA?
ALCOA is the acronym for a framework to ensure data integrity in GxP (GMP/GDP) regulated pharmaceutical environment. It outlines five attributes that should be met in all GMP records – on paper or electronically.
ALCOA is an acronym for Attributable, Legible, Contemporaneous, Original, and Accurate.
The US FDA started to adopt it in the 1990s as a common basis for the assessment of the trustworthiness of a record by the manufacturer, the laboratory, or the inspector.
What Is ALCOA+?
ALCOA+ is the extended version of ALCOA. It adds four additional attributes to deal with electronic data management throughout its lifecycle.
The original five principles could no longer be applied when paper was replaced by digital systems in the pharmaceutical manufacturing industry. ALCOA+ is the answer to that.
There are four more attributes: Complete, Consistent, Enduring, and Available.
ALCOA+ is now the minimum requirement for FDA, EMA, MHRA, WHO, and PIC/S for GMP Data Integrity compliance.
What Is ALCOA++?
The most advanced form of ALCOA++ is the model. It introduces five new attributes specifically for use in complex digital environments, such as AI-driven systems, cloud solutions, and manufacturing integration networks.
The five other attributes are Integrity, Robustness, Transparency, Accountability, and Reliability.
The new Annex 22 for AI systems and the draft EU GMP Chapter 4 are the first comprehensive regulatory documentation to formally recognize ALCOA++ as the standard for all GxP data – paper, electronic and hybrid.
Principles of ALCOA, ALCOA+, and ALCOA++ with Examples and illustrations:
ALCOA (5 Principles) with Examples and Illustrations:
1. Attributable:
Definition: The collected data must be attributed to who performs the action and when, if a record is changed, who did it and why?
Example:
When conducting validation, the test results must be dated, and the initial validation should be done by the person involved in conducting the test. If there is any change in the monitoring system, the details of the change should be in the audit trail, and any corrections made by the person should be recorded and dated. A signature log must be maintained for the identification of initials and the person who completed the paper record.

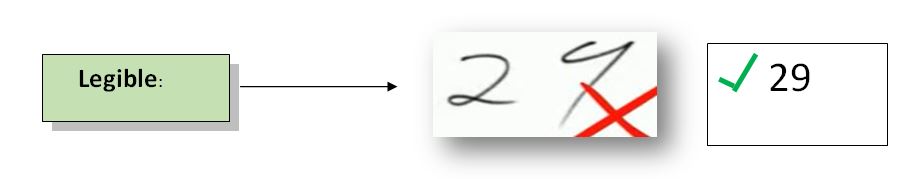
2. Legible:
Definition: Data must be recorded permanently in a durable record medium and easy to read. If data records are permanent and readable, they can be easily assessed through the entire product data lifecycle. It helps in the easy retrieval of all previous data if needed.
3. Contemporaneous:
Definition: The data should be recorded at the time and date of work performed. The timestamp should we follow in order.
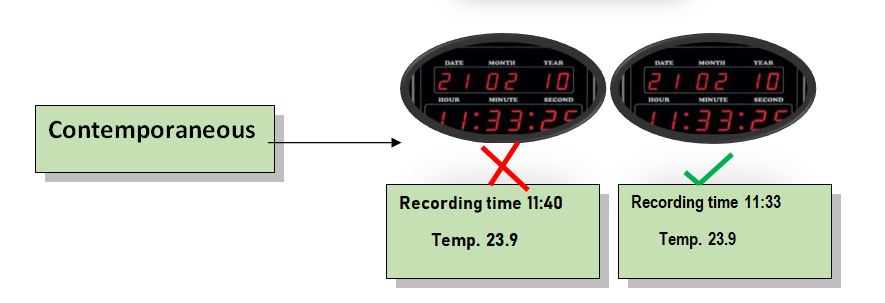
Example:
When conducting validation protocol, the result of the test performed should be recorded in an online sequence. Recording the results should be dated with a timestamp and then logged into the electronic system.
4. Original:
Definition: The information must be recorded as original or in a certified true or original copy; this may be an acceptable protocol, a database, or a notebook.
Example:
For example validation test is being recorded on a given protocol because recording test results in a Notebook may be a chance of error. If the original data is handwritten, it must be stored in an electronic system.
5. Accurate:
Definition: No error or editing was performed without documented amendments to ensure the accuracy of the data and records. The data must have the following characteristics.
- It should be complete
- It should be free from error
- It should be e reflective of the observation
Any change made to the recorded data should be documented to ensure the accuracy of the data collected. Follow security checkups in the electronic system. Ensure there is a witness to check for any critical data collection.

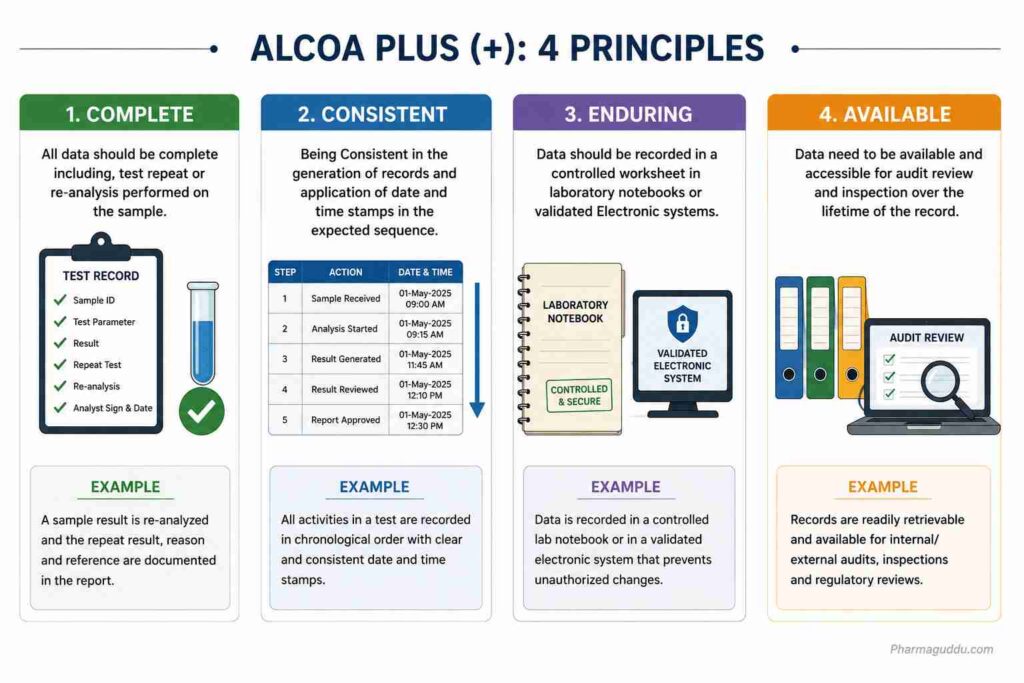
ALCOA PLUS (+): (4 Principles)
6. Complete:
Definition: All data should be complete including, test repeat or re-analysis performed on the sample.
7. Consistent:
Definition: Being Consistent in the generation of records and application of date and time stamps in the expected sequence.
8. Enduring:
Definition: Data should be recorded in a controlled worksheet in laboratory notebooks or invalidated Electronic systems.
9. Available:
Definition: Data need to be available and accessible for audit review and inspection over the lifetime of the record.
ALCOA++: (1 Principle)
10. Traceable (Transparency)
Definition:
Traceable is data, action, change, and record able to be traced back to their source throughout their lifecycle. A clear “audit trail” indicating who was responsible for an action, what action was taken, when and why.
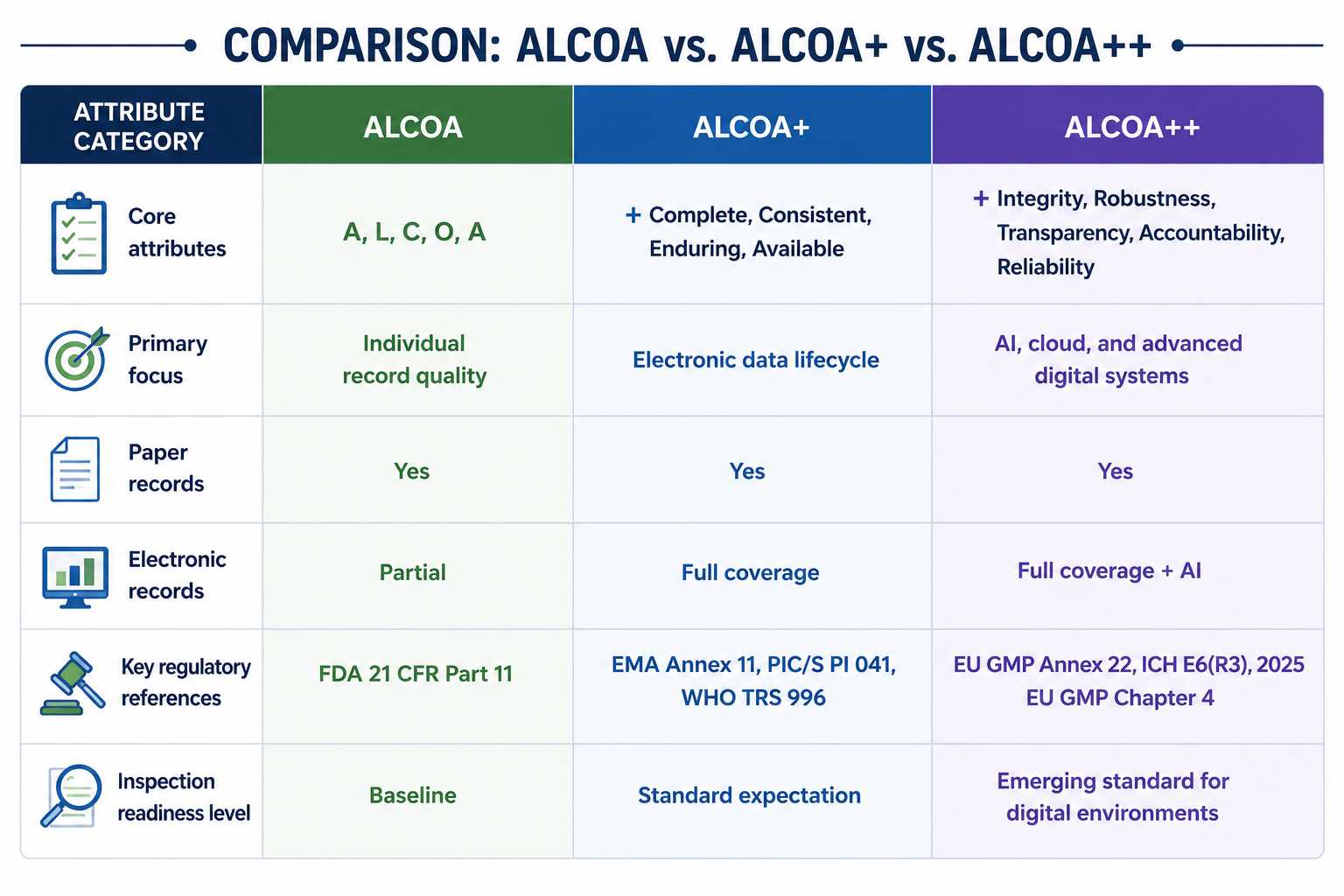
Comparison Table: ALCOA vs. ALCOA+ vs. ALCOA++
| Attribute Category | ALCOA | ALCOA+ | ALCOA++ |
| Attributes | Attributable, Legible, Contemporaneous, Original, and Accurate | +Complete, Consistent, Enduring, Available | +Integrity, Robustness, Transparency, Accountability, Reliability |
| Focus On | Quality Records of individuals | Focusing on electronics data records | Focusing on AI, cloud, and advanced digital systems |
| Electronic records | Patial | Full coverage | Full coverage+AI |
| Paper Records | Yes | Yes | Yes |
| Key regulatory references | FDA 21 CFR Part 11 | EMA Annex 11, PIC/S PI 041, WHO TRS 996 | EU GMP Annex 22, ICH E6(R3), 2025 EU GMP Chapter 4 |
Common ALCOA Mistakes Found During GMP Inspections
ALCOA Mistakes, which are regularly seen during GMP inspections, are provided below.
- Having more than one staff member access a system using a single staff account. It’s one of the top three findings for FDA, EMA and MHRA inspections.
- The range of data integrity concerns, from incomplete audit trails to poor access control, remains constant and is still being uncovered by regulators. If an audit trail is implemented but there is no review procedure, it is considered a system control gap.
- Use of fluid ink to make data correct is another finding that hides the original entry in documents. Draw one line and write the correction, sign, date and state the reason (But it should be justifiable). The only way is to do that.
- Backdating Entries: In at least one FDA case, the operators had altered and destroyed original batch records and replaced them with backdated pages. This is fraud and the most serious regulatory implications.
- Discarding Raw Data: One of the common findings is that only the summary report is retained and the raw chromatogram is discarded, thus making verification of original data impossible. Raw data shall be kept for the entire retention time.
- Pre-Recording of Data: Making a batch record step but not doing the activity results in a falsified contemporaneous record. The intent doesn’t matter; it’s considered falsification.
- Paper records should be signed and blank fields should be marked N/A. The blank space is unsigned and raises the question as to whether it was added or removed after the fact.
Case Study: Unsigned Batch Manufacturing Record — FDA Inspection Finding (2022)
Background:
A pharmaceutical company was producing a product in a batch process when an operator incorrectly completed a Batch Manufacturing Record (BMR). The operator crossed out the incorrect entry (the right GMP procedure) but provided no signature, date, time or reason for the correction. The BMR was reviewed and approved via the standard quality workflow process without anyone picking up the gap.
Finding by FDA Investigator:
As part of their planned FDA audit inspection, investigators asked to review a number of historical BMR records. On examining the records, they identified the unsigned correction. One strikes through was placed over the original value in the entry but no identity, no date, no time, no reason was attached.
The investigator made this an observation on data integrity and specifically requested assurance from the company that it was in compliance. The issue was simple: a change without a signature, date, time and reason is undetectable, so it cannot be known when the change was made, or if it was made before or after batch release, or even by whom it was made. Both the original error and the correction are questionable.
ALCOA Principles Violated:
- Attributable — The correction is able to be attributed to a specific person.
- Contemporaneous – No date and time – there is no evidence that the correction was made at the time of the activity.
- Accurate — Record cannot be fully trusted due to an unknown context of the correction.
Root Cause:
The operator was familiar with the procedure for making a strike-through an error, but he did not do it completely. This indicates a training gap, that is, knowing that a correction has to be struck through, but not knowing that the correction is only GMP-compliant if it has a signature, date, time and reason.
The second fault was in the process of review. It was not evident to either the batch reviewer or the QA approver that the correction was not complete prior to the release of the BMR. As a result, it appears that error correction needs were not in the formal BMR review checklist.
How to handle this data integrity issues- Best Approach
- Cross out 1 wrong answer with a horizontal line. The original value must be legible.
- Write the correct value immediately next to or above it.
- Sign with your name or initials.
- Put Date and Time of the correction.
- Enter a short reason, such as: Incorrect batch number entered – changed to actual batch number used.
Corrective and Preventive actions implemented:
Immediate Actions:
- Specific BMR was fully reviewed to determine impact of the unsigned correction on batch quality and disposition. A formal impact assessment was documented.
- Deviation issued by QA to capture the finding and investigation.
Corrective Actions:
- As a lookback exercise, all BMRs from the last year have been checked for similar unsigned / incomplete corrections.
- An FDA response was provided that included formal documentation of corrective actions.
Preventive Actions:
- The BMR reviewer checklist was amended to provide a specific line item: “All corrections signed, dated, timed and reason stated – verified”.
- Training was carried out on the GMP error correction requirements for all production & QA staff.
- Effectiveness of training was measured by a practical exercise: operators were provided with a BMR containing a deliberate error and asked to rectify these errors correctly under observation.
- SOP was updated with examples of proper and improper ways to correct errors.
Risk-Based Approach to ALCOA++ Compliance
There are different levels of regulatory risk for data. A Risk Based Approach focuses your resources on the right areas.
Critical Risk Data (Maximum ALCOA++ Controls Required): Data that could have a significant impact on the project if it were not available.
- Batch manufacturing records
- Results from the laboratory tests and raw chromatograms
- Stability study data
- Environmental monitoring records
- OOS investigation records
High-Risk Data
- Equipment calibration logs
- Cleaning verification records
- Documentation of deviation and CAPA
Medium-Risk Data
- Training records
- Supplier audit reports
- Change control documentation
Lower-Risk Data
- Internal meeting minutes
- Non-GMP administrative records
Conclusion
Recently data integrity issues are rising more and more on the daily basis, because Regulatory bodies are more focused on real data, by use of more and more tools and systems, it gets more complicated since the impact of data integrity is paying off high cost. It is something that has a high priority.
Frequently Asked Questions (FAQs):
Ans. There is a total of 10 principles for ALCOA (5), ALCOA+(4), and ALCOA++(1)
Ans. Attributable, Legible, Contemporaneous, Original, and Accurate.
Ans. Complete, Consistent, Enduring, and Available
Ans. No, Data integrity is good, but Data integrity issues are bad for any Pharmaceutical industry.
Ans. EU GMP Annex 22 is the first EU regulatory document specifically created for AI-based systems in the pharmaceutical manufacturing sector. It reaffirms the importance of the ALCOA++ principles for all processes involving AI assistance and provides clear guidelines for validation, consistency of performance, and change management. It lays out a requirement for human oversight of AI models. Generative AI and large language models are not included, but may be added in the future.
Ans. Cross out the wrong entry with one line, leaving the original entry clear. Write the correct entry next to it. Write your name or initials, date and Reason for why the correction was made. Do not use correction fluid, correction tape or other means of concealing the original.
Ans. Commonly reported errors are: shared system logins that make it difficult to attribute, illegible or overwritten Logbooks, and no audit trail reviews, or raw chromatograms discarded, or manual editing without trace in a validated system. Pre-record or backdated records are considered the most serious and normally are subject to warning letters not 483 observations.
Related Articles around to FDA and Guidelines:
- FDA Form 483 | Warning Letters: How to Handle, Form, Example
- Good Documentation Practice (GDP) in Pharmaceuticals
- CAPA Corrective and Preventive Action in Pharmaceutical
- Pharmaceutical Quality System (PQS), ICH Q10 Guidelines
- Change Management System in Pharmaceuticals
- Complete Overview of ISO CleanRoom Classification and Risk Assessment
- Difference Between Classified and Non-Classified Areas in Pharmaceuticals
- Good Laboratory Practice (GLP) in Pharmaceutical
- What is GMP | cGMP | GMP Principle
- ICH Guidelines in Pharmaceutical (updated)
- GxP in Pharmaceuticals industries (FDA guidelines)
Related useful references
- FDA. Data Integrity and Compliance With Drug CGMP: Questions and Answers Guidance for Industry (2018). Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/data-integrity-and-compliance-drug-cgmp-questions-and-answers-guidance-industry
- EMA. Reflection Paper on Data Integrity (2018). Available at: https://www.ema.europa.eu/
- WHO. Guidance on Good Data and Record Management Practices, TRS 996, Annex 5 (2021). Available at: https://www.who.int/
- MHRA. GxP Data Integrity Guidance and Definitions, Revision 1 (2018). Available at: https://www.gov.uk/government/publications/gxp-data-integrity-guidance-and-definitions
- PIC/S. PI 041-1: Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments (2021). Available at: https://picscheme.org/
- ICH. E6(R3): Good Clinical Practice Guideline (2025). Available at: https://www.ich.org/
- European Commission. EudraLex Volume 4 – EU GMP Guidelines. Available at: https://health.ec.europa.eu/medicinal-products/eudralex/eudralex-volume-4_en
- FDA. 21 CFR Part 11: Electronic Records; Electronic Signatures. Available at: https://www.ecfr.gov/
- ISPE. Pharmaceutical Engineering Magazine. Available at: https://ispe.org/pharmaceutical-engineering
- IntuitionLabs. ALCOA+ Principles: A Guide to GxP Data Integrity. Available at: https://intuitionlabs.ai/alcoa-principles-guide-gxp-data-integrity/

Naresh Bhakar is the Founder and Author at Pharmaguddu.com, bringing his extensive expertise in the field of pharmaceuticals to readers worldwide. He has experience in Pharma manufacturing and has worked with top Pharmaceuticals. He has rich knowledge and provides valuable insights and data through his articles and content on Pharmaguddu.com. For further inquiries or collaborations, please don’t hesitate to reach out via email at [email protected].