
GMP & Pharmaceutical Manufacturing Expert
✓ Reviewed by: Pankaj Sharma - Quality Control Specialist
Reviewed for Quality Control accuracy, laboratory practices, analytical methods, and technical relevance
📅 Last Updated: June 7, 2026
Cleaning method validation (sometimes referred to as cleaning process validation) involves documenting assurance that a specific cleaning procedure will consistently lower residues of a previous product, cleaning agent, or microbial contaminants on pharmaceutical manufacturing equipment to an acceptable level.

Pharmaceutical products are generally contaminated by microorganisms, cleaning agents, or other materials, but in many cases, it may be due to the repeated use of the same equipment for processing. To avoid this problem FDA introduced the Cleaning Method Validation process for pharmaceutical industry.
The cleaning procedure must be strictly followed to execute the cleaning method. Normally, only cleaning systems for product contact surfaces need to be validated. But now, Consideration should be given to non-contact parts where the product may be migrated.
Cleaning procedures for product changeover in non-dedicated multi-purpose equipment should be fully validated.
For Cleaning method validation FDA says there must be a standard procedure and written documentation at every stage. The procedure includes the concentration of the detergents that should be in place for each piece of equipment, whether or not the CIP (clean in place) system is used to clean processing equipment, and whether microbial aspects of cleaning should be considered.
After the cleaning process, equipment may be subjected to sterilization or sanitization procedure, depending on whether products may support microbial growth.
FDA says, that direct surface sampling and rinse sampling procedures are used for samples for cleaning method validation.
Cleaning Method Validation History:
The Food and Drug Administration states that instrumentation is to be cleaned before use, which is nothing new, hence in 1963, GMP rules (Part 133.4) declared as follows, “Equipment shall be maintained in a very clean and orderly manner”. A similar section on instrumentation cleansing (211.67) was enclosed within the 1978 CGMP rules.
Of course, the principle for requiring clean instrumentation is to stop contamination or adulteration of the drug products. Historically, the FDA has investigated inadequate cleaning and maintenance of equipment with poor dust control systems. Also, historically speaking, the FDA found Product contamination, or the cross-contamination of drug products with potent steroids or hormones, as a number of the products recalled over the past decade because of actual or potential penicillin cross-contamination.
Food and Drug Administration in one event augmented awareness of the potential cross-contamination because inadequate procedures were followed, resulting in the recall of a finished drug product, eg. Cholestyramine Resin USP in 1988.
Contamination due to Solvents:
The bulk pharmaceutical chemical wants to manufacture the drugs, but has been contaminated by intermediates on a small scale. In this case, the cross-contamination is believed to have been due to the reuse of recovered solvents. The recovered solvents had been contaminated due to a lack of control over the reuse of solvent drums.
Drums that had been used to store recovered solvents from a chemical production method were later won’t to store recovered solvents. The company didn’t have controls over these solvent containers, didn’t do adequate testing of container solvents, and did not have validated cleaning procedures.
Contamination During Shipments:
Some chemical shipments may contaminate bulk pharmaceuticals, resulting in contamination that is utilized in the facility, Likes fluid bed dryers may be contaminated with chemicals, leading to cross-contamination in batch lots made at that site, where no pesticides were normally produced.
FDA instituted an import alert in 1992 on a foreign bulk pharmaceutical manufacturer that company-made a potent steroid product in addition to a non-steroidal product using common equipment. At that time FDA considered the potential for cross-contamination to be vital and create a serious health risk to the general public.
The firm had recently started a cleaning validation program at the time of the examination, and it had been found inadequate by the Food and Drug Administration. The explanation was that the firm was only searching for proof of the absence of the previous compound, but from TLC tests on the rinse water, the firm had evidence of the presence of residues of reaction byproducts and degradants of the prior process.
Reference: fda.gov/validation-cleaning-processes-793
General Requirements for Cleaning Method Validation:
The FDA expects corporations to have their own written procedures (SOPs) and descriptions for cleaning processes that have been used for various apparatus. Suppose corporations are using one cleaning method for cleaning between different batches of an equivalent product and use a unique way to clean product equipment. In that case, we tend to expect the written procedures to address these different scenarios.
Normally only cleaning procedures for product contact surfaces need to be validated. But as of now, Consideration shall be given to non-contact parts where there is a chance of product migration, e.g., mixing shaft, seals, flanges, fans, and oven heating elements. Fluid bed drier (FBD) bags are another example of a difficult-to-wash apparatus and are usually dedicated to a selected product.
When the cleaning process is used only between batches of the same products or lots of the same intermediates in the bulk process, like a single product line, in this case, only visible clean criteria should be met. After cleaning, equipment may be subject to sterilization and sanitation, which may support microbial growth.
Cleaning Method Validation Approach:
Cleaning validation can be done for specific products or representatives of product groups.
- Product-specific cleaning validation: There must be established documented evidence for product-specific validation with the same active principle. The use of the cleaning agent must be done based on a preplanned validation protocol.
- Cleaning validation of product families or groups of risk. The selection of similar products and processes concerned range is considered acceptable. A single validation study of the method can be carried out, which considers the relevant criteria, called “Grouping.” The following are the “grouping” criteria.
- The same dosage forms
- Similar processes
- Similar formulations
- The Physiochemical properties of active ingredients, e.g., %solubility and lipophilicity.
- Similar therapeutic potency
- Highly toxic products
- Revalidation (control of change to a validated cleaning method is required.
Criteria for Revalidation
- Revalidation shall be carried out In case of equipment, product composition, or cleaning process change.
- Periodic revalidation must be carried out as per a given interval, such as yearly or every two years.
Documentation for Cleaning Validation:
Validation Protocol:
The validation protocol must be ready for cleaning validation before starting the validation activity. The following points must be considered for this protocol.
- Objective and scope
- Responsibility
- Product composition ( active ingredient and main components of detergents used for cleaning) will be tested.
- Description of the manufacturing method
- The time interval between production end and the start of cleaning
- Details of cleaning validation procedure
- Sampling position, areas, and diagram with difficult-to-clean area
- Sampling procedure
- Acceptance criteria
- The analytical method includes a limit of detection and a limit of quantitation of those methods.
- The protocol should be a formal approach by the responsible and authorized person.
The Following committee is responsible for the Validation Protocol:
- Validation Specialist – Who can write and coordinate the procedure?
- Manufacturing – Who can write SOPs and provide training to a person involved in the activity?
- Quality Assurance/Control – For Approval and implements analytical methods
- Engineering– Communicates about equipment data changes.
- R&D – Performs studies on recovery, methods validation, transfer methods, and selects new cleaners.
Validation Report:
After cleaning validation completion, prepare the report, and the conclusion of this report should state if the cleaning process has been validated successfully. Validation reports shall consist of:
- The validation protocol.
- Related results and acceptance limits.
- Result of the validation of the analytical method.
- Any deviation from the manufacturing or cleaning process.
- The person involved in a validation study, including the name of the person who sampled the swab.
- The period during which validation was performed.
- Summary of the results.
- The report should be approved.
- Validation certificates: at the end, validation certificates shall be prepared and signed.
Sampling Method for Cleaning Validation:
The following two types of sampling methods are given below:
- Direct surface sampling:
- Rinse sampling:
1. Direct surface sampling:
- For direct sampling, take two pieces of cotton wool of 1 gm each and divide them into two parts equally.
- Fix the stencils on the plain equipment surface according to the validation protocol. Avoid stencils. If sampling is not possible anywhere with stencils, e.g., small parts of the uneven surface, the pieces should be swabbed completely without a stencil.
- Dip the swab with the artery clamp into the solvent, and press the swabbing unit until it drops free.
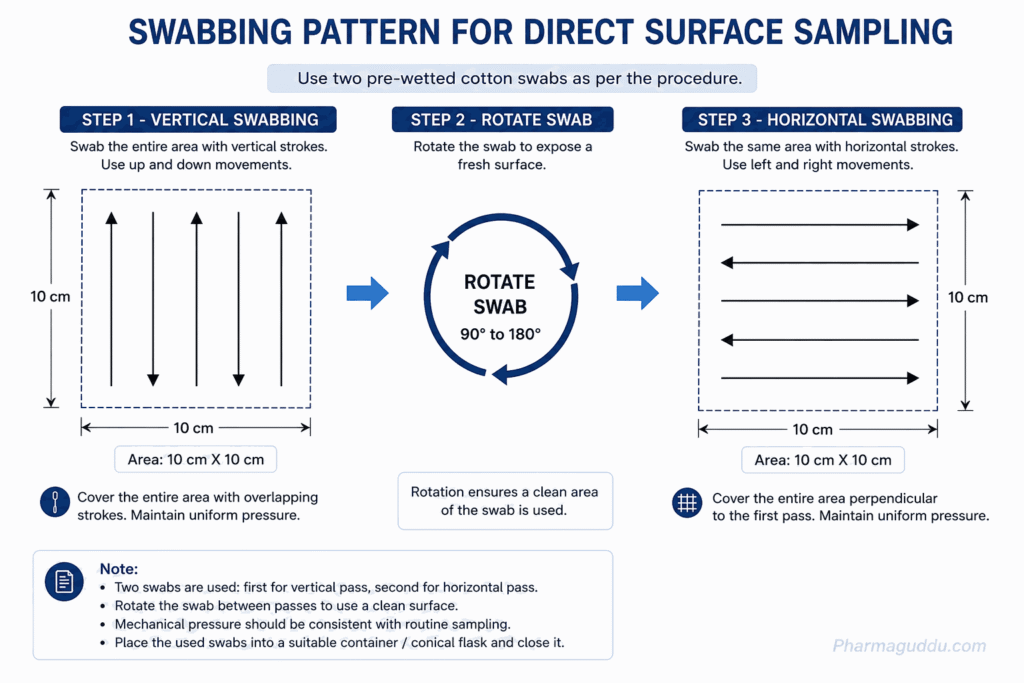
Swab the inner area of the stencil with the artery clamp. Lead the first piece of the surgical cotton wool with smooth pressure with small weight-wise moves from one stencil corner to the diagonally opposite corner. Like that, Swab with the second piece of cotton wool in the opposite direction. The second stencil has to be sampled in the same manner. The mechanical pressure applied by the chemist while sampling should be the same as during routine sampling. Now, put the pieces of cotton wool into a conical flask and close it.
2. Rinse Sampling:
A rinse sample is used where a large area or part of the equipment could not be possible with a swab. It should be rinsed, sampled, or directly extracted by solvents. For example, Tubes, nozzles, pipes, or container surfaces that are not accessible for direct surface sampling have to be rinsed with a solvent. The compound that is to be sampled should be freely soluble in the solvents. For example, for FBD Filter bags, soak filter bags in solvent and sake for 2 hours. Evaporate 1 liter of rinse sample to dryness by rotary evaporation.
Sample Preparation for Cleaning Validation:
Swab sample: the sample should be taken according to the procedure below or by using other validation methods. Shake the cotton wool 5-6 times with 200 ml of solvent for 30 seconds. Remove the solvent after each shaking step, press out the cotton wool with a spatula, and evaporate the collected extracts until dryness; by rotary evaporation.
Dissolve the residue in the required quantity of solvent and transfer it to the volumetric flask. Filter the solution and determine the amount of the residue according to the method specified in the validation protocol. Keep in mind that the standard used for calculation has a concentration that is similar to the sample.
Rinse sampling: evaporated the collected extracts until dry by rotary evaporation. Dissolve the residue in the required quantity of solvent and transfer it to the volumetric flask.
Filter the solution and determine the amount of residue according to the method specified in the validation protocol. Keep in mind that the standard used for calculation has a concentration that is similar to the sample. Always analyze a blank solution in parallel and the solvent handled in the same manner as the swab sample.
Validation of Analytical method:
Active ingredients and detergents shall be soluble in the sampling solvent. Before the cleaning method validation, the analytical method has to be validated.
- Sampling material: Keep in mind that cotton wool, polyurethane foam (PUF), or GFC filter, sampling material shall not interfere with the analysis. Recovery from sampling material shall be more than 70%, otherwise, modify the solvent or sampling material.
- Swab material: surgical cotton wool (50% cotton,50% viscose), artery clamp, solvent (for stencil) made up of polyethylene foil, sampled area of each equipment with plain surface should be 800 cm square (2 stencils of 400 cm square), 50 ml conical flask with stopper, volumetric flask with stopper.
- A few of the analytical methods for the analysis of cleaning validation samples are:
- HPLC
- GC
- HPTLC
- TOC
- UV Spectroscopy
- pH
- conductivity
- ELISA
These methods can be used alone or in combination, depending upon the analysis required.
Limits establishments for cleaning validation: The limits selected depend upon the materials involved and their therapeutic dose.
ACCEPTANCE CRITERIA for Cleaning Validation:
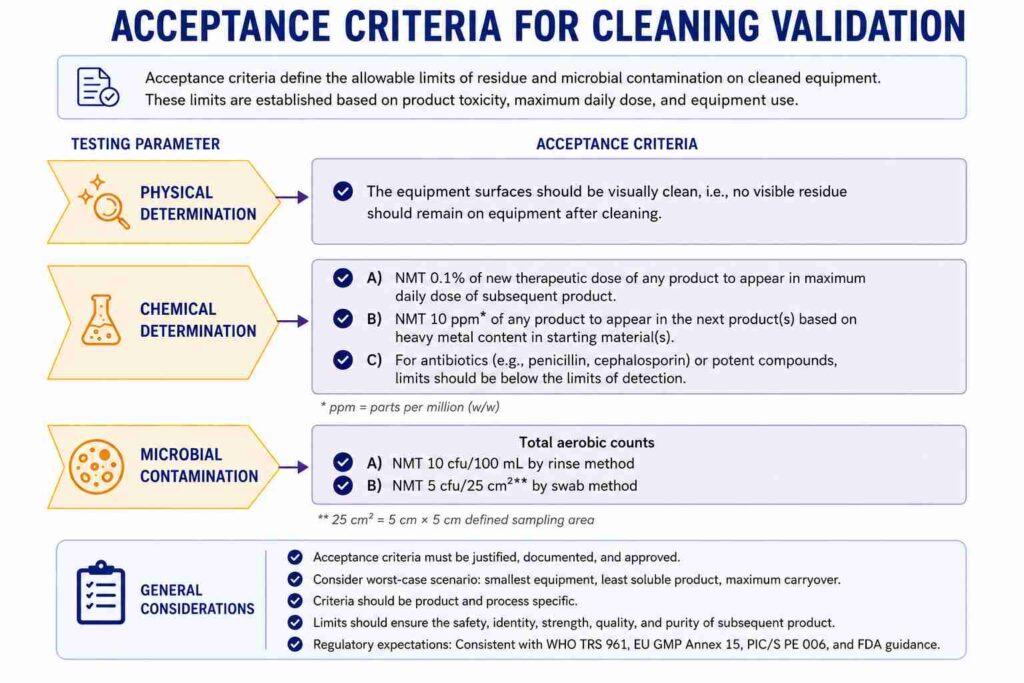
CONCLUSION:
According to the above article, cleaning validation is a documented process that is highly effective in proving efficiency and consistency in the pharmaceutical sampling and cleaning of equipment. Therefore, it is crucial to have an effective cleaning method in place to meet regulatory requirements.
References
- U.S. Food and Drug Administration. 21 CFR Part 211, Section 211.67 — Equipment Cleaning and Maintenance. FDA.gov.
- U.S. Food and Drug Administration. Guide to Inspections of Validation of Cleaning Processes. FDA, 1993.
- European Medicines Agency. Guideline on Setting Health Based Exposure Limits for Use in Risk Identification in the Manufacture of Different Medicinal Products in Shared Facilities. EMA/CHMP/CVMP/SWP/169430/2012.
- World Health Organization. Technical Report Series 1033, Annex 2: Points to Consider When Including Health-Based Exposure Limits in Cleaning Validation. WHO, 2021.
- International Society for Pharmaceutical Engineering (ISPE). Guide to Cleaning Validation Lifecycle: Applications, Methods, and Controls. ISPE, 2020.
- ICH Q7. Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients. ICH, 2000.
- PDA Technical Report No. 29. Points to Consider for Cleaning Validation. Revised 2012.
- FDA Warning Letter, CMS 672951 (2024). Drug Manufacturer — Inadequate Cleaning Validation for Multipurpose Equipment.
- Leucine.io. 2025 Cleaning Validation Guidelines: FDA, EMA, MHRA & WHO. Updated September 2025.
- NCBI/PMC. Evaluation of Swab and Rinse Sampling Procedures and Recovery Rate Determination in Cleaning Validation. PMC7758009.

Naresh Bhakar is the Founder and Author at Pharmaguddu.com, bringing his extensive expertise in the field of pharmaceuticals to readers worldwide. He has experience in Pharma manufacturing and has worked with top Pharmaceuticals. He has rich knowledge and provides valuable insights and data through his articles and content on Pharmaguddu.com. For further inquiries or collaborations, please don’t hesitate to reach out via email at [email protected].