Quality Control Specialist | Industry Experience
✓ Reviewed by: Pankaj Sharma - Quality Control Specialist
Reviewed for Quality Control accuracy, laboratory practices, analytical methods, and technical relevance
📅 Last Updated: March 23, 2024
Learn About Standard Operating Procedure for Handling of Deviations in pharmaceuticals | Types of deviations | Deviation Flow chart | Devations Guidelines | Deviation Numbering | Deviation management.

1.0 OBJECTIVE:
1.1 To lay down that deviations from manufacturing instructions and quality-related procedures are recorded, investigated, and evaluated for their impact on product quality in a manner consistent with Good Manufacturing Practices, and that any action arising from the review is formally recorded and progressed.
2.0 SCOPE:
2.1 This procedure is applicable to all related user departments at Pharmaceuticals.
3.0 REFERENCE:
3.1 ICH Guideline
4.0 RESPONSIBILITY:
4.1 Originator
4.1.1 To Identify and raise the deviation.
4.1.2 To inform to department head and QA about the deviation
4.1.3 To Provide the required information and assist in the investigation process.
4.2 Department Head / Designee
4.2.1 To Review the raised deviation report.
4.2.2 To Carry out investigation & root cause analysis.
4.2.3 To Propose the Appropriate Corrective and Preventive Action.
4.2.4 To Ensure implementation of proposed CAPA.
4.3 Head QA
4.3.1 To Review the investigation report and root cause.
4.3.2 To Do the impact analysis of the deviation.
4.3.3 To Ensure proper Corrective Action and Preventive Action are taken.
4.3.4 To Ensure the implementation of CAPA.
4.3.5 To Ensure the adequate closure of deviation & Monitoring of CAPA.
5.0 ACCOUNTABILITY:
5.1 Head-Quality Assurance.
6.0 DEFINITION:
6.1 A deviation is defined as a departure from an approved written procedure/instructions / established standards of quality system in the organization.
6.1.1 Planned Deviation: Any deviation from established and approved procedure opted deliberately for the temporary time period prior to the execution of an activity without affecting the quality and safety of drug substances or drug products shall be termed as planned deviation.
The planned deviation is restrained to only 3-time changes for the particular system, document, or process. If the same planned deviation is repeated, then preventive actions shall be reviewed and if required, changes in the system, process, or document can be done as per the procedure of “Change Control”.
6.1.2 Unplanned Deviation: Any deviation that occurred accidentally during or after the execution of an activity due to system failure, equipment breakdown, or manual error shall be termed as unplanned deviation.
6.2 Documents covered under the scope of this standard operating procedure include the following, but are not limited to:
6.2.1 Standard Operating Procedures
6.2.2 Manufacturing Procedures
6.2.3 Packaging Procedures
6.2.4 Cleaning procedure
6.2.5 Environmental monitoring
6.2.6 Batch analysis (In-process Control)
6.2.7 Quality Control Testing
6.2.8 Utility operations
6.2.9 Calibration procedures
6.2.10 Analytical procedures
6.2.11 Batch release
6.3 No process deviation shall be permitted under normal operating conditions; however, in case the deviation is unavoidable, the same should be immediately brought to the notice of QA Head.
6.4 No deviation shall be permitted from Pharmacopoeia and other Regulatory specifications.
6.5 Deviation can be categorized into three categories – Critical, Major & Minor.
6.5.1 Critical Deviation: Deviation that has an immediate & direct impact on the quality, purity, strength, stability, and safety of the drug product. For example: manufacturing instructions not followed, SOPs/STPs/Specifications not followed, etc.
6.5.2 Major Deviation: Deviation that may have a direct or indirect impact on the quality, purity, strength, stability, and safety of the drug product. For example: Machine breakdown during processing, mix-up of cartons of same product with different strengths, etc.
6.5.3 Minor Deviation: Deviation that does not have a direct or indirect impact on the quality, purity, strength, stability, and safety of the drug product. For example, Line clearance not taken, spillage of material during dispensing, etc.
6.6 A person coming across any deviation shall immediately report to the department head/designee and QA. Based on the nature of deviation, concerned department head/QA Head shall suggest an immediate action or stoppage of activity and further action to be taken and deviation shall be reported as per Format No. F/QA/004/B/05.
6.7 The timeline for logging of deviation is within 24 hours after its occurrence.
6.8 The initiator shall collect deviation forms from QA. QA representative shall issue the deviation form after assigning the deviation number as follows:
| Originating Department | Deviation Sequential No from “001” to “999” | Slash | Year from “00” for year “2000” to 99 for year 2099 |
| ( ___ ___ ) | (___ ___ ___) | ( / ) | ( ___ ___ ) |
6.8.1 The first two characters are alphabets, which shall identify the department code as mentioned under SOP on SOP.
6.8.2 3rd Character is Alphabet “D” stands for Deviation.
6.8.3 4th, 5th, and 6th characters shall indicate the serial number for deviation report.
Example: 001 for 1st Deviation
6.8.4 7th Character is “ / ”.
6.8.5 The 8th and 9th characters represent the last two digits of the financial year.
Example: 24 for the year 2024
6.9 The initiator shall indicate the type of deviation and mention the description of deviation along with additional details regarding the product, batch number, etc. in the deviation report.
6.10 The initiating department head/designee shall review the deviation form and discuss it with a person involved in the activity and probable reason for the deviation shall be provided.
6.11 The report shall then be forwarded to the concerned department head for their review and comments, as applicable, based upon the nature of deviation.
6.12 Investigation to be carried out by the initiating department head along with other cross-functional department heads to identify the root cause. The following tools shall be used for root cause analysis:
6.12.1 Brainstorming
6.12.2 Five Why’s Analysis
6.12.3 Ishikawa diagram (Fishbone diagram / 6M technology)
6.13 Investigation shall include (but not limited to):
6.13.1 Review of Batch Manufacturing Record / Batch Packing Record
6.13.2 Equipment Usage Log Review
6.13.3 Analytical document (if applicable)
6.13.4 Breakdown history of equipment/utility system
6.13.5 Discussion with the person involved in activity
6.13.6 Review of relevant SOPs / STPs / Specifications
6.14 Based on identified root cause from the investigation, the department head shall propose an appropriate CAPA. The same shall be recorded in deviation form and CAPA form, along with the CAPA reference Number in the Deviation form.
6.15 The deviation form along with investigation report shall then be submitted to QA Head for review and evaluation.
6.16 QA Head shall evaluate the deviation report with respect to the impact on other batches/products, yield, product quality, process performance, etc.
6.17 In case of deviation related to the products of contract giver, the deviation shall be immediately brought to the notice of qualified person of contract giver through e-mail, telephonically, or any other means.
6.18 After reviewing details like root cause identified, CAPA, and contract giver comments/approval (wherever applicable); the QA Head shall recommend the necessary actions and give final approval for the deviation.
6.19 In case, a deviation is rejected, then inform the initiator department and the deviation shall be closed with proper justification for rejection.
6.20 Details shall be recorded in the Deviation register.
6.21 QA shall retain the original for records irrespective of the approved/rejected status of the deviation report, and a photocopy of the same shall be filed with the subjected batch manufacturing/packing record or analytical reports (as applicable).
6.22 After approval, the initiating department head shall implement the CAPA as proposed and provide the supporting document to QA for review.
6.23 QA Head / Designee shall verify the satisfactory implementation of CAPA and closure of deviation shall be done.
6.24 In case of batch/material specific deviation, batch/material shall be released after closure of the deviation.
6.25 Deviations shall be closed within 45 working days and for critical and major it shall be 30 days after initiation of deviation. If any deviation extends the timelines of 45 days /30 days then justification for delay and progress report are to be documented as per the “Justification form for the delay in closure of deviation”.
6.26 Deviations arising out of testing shall be handled according to the procedure of “Handling Out of Specification”.
6.27 Trending analysis of all deviations shall be carried out on half yearly basis to assess the reoccurrence of deviations.
6.28 Any amendments to systems, documents, or procedures arising out of the recommendations for CAPA shall be done according to the procedure of “Change Control”.
7.0 ABBREVIATIONS:
7.1 QA: Quality Assurance
7.2 SOP: Standard Operating Procedure
7.3 STP: Standard Testing Procedure
7.4 CAPA: Corrective Action and Preventive Action
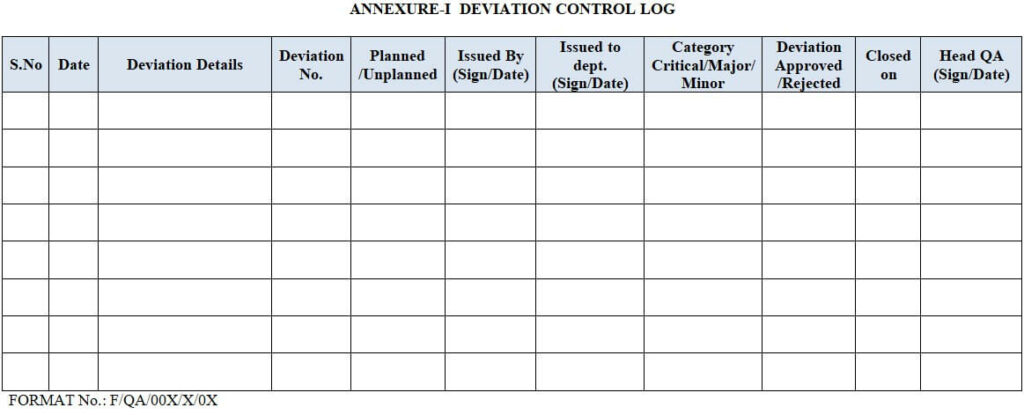
8.0 ANNEXURE for Handling of Deviations:
| Annexure-I | Deviation Control Log |
| Annexure-II | Deviation Control Form |
| Annexure-III | Deviation Flow Chart |
| Annexure-IV | Justification form for delay in closure of deviation |
END OF DOCUMENT
| SUPERCEDE SOP NO. | CCR NO. | REASON FOR REVISION |
| — | — | Write the Reason for Changes |

Pankaj Sharma — Quality Control Specialist
Pankaj Sharma is a pharmaceutical QC professional who reviews our technical content for GMP accuracy, laboratory practices, and regulatory relevance.