GMP & Pharmaceutical Manufacturing Expert
✓ Reviewed by: Pankaj Sharma - Quality Control Specialist
Reviewed for Quality Control accuracy, laboratory practices, analytical methods, and technical relevance
📅 Last Updated: July 9, 2023
Schedule M was incorporated by the Drug and cosmetic act of 1940 in 1988. Schedule M is also known for Good manufacturing practices. But earlier there were no rules and regulations that dictated us to know, How is our pharmaceutical building, location, premises, water system, construction area, warehouse, etc.
Introduction to M Schedule
Schedule M is a part of the Drugs and Cosmetics Act of India, 1940. Schedule M has its own requirements for the manufacture, sale, and distribution of drugs in pharmaceuticals. Schedule M sets the minimum standards for GMP that all pharmaceutical manufacturers in India must follow. Schedule M for GMP covers all aspects of pharmaceutical manufacturing, including facilities, equipment, personnel, documentation, and quality control.
Objectives of Schedule M
The objective of Schedule M for GMP is to ensure that pharmaceutical manufactured products are safe, effective, and of high quality. It achieves this objective by setting minimum standards for GMP and requiring pharmaceutical manufacturers to comply with these standards. The other objectives of Schedule M for GMP are to:
- Ensure that the manufacturing process is well-documented, and all records are maintained
- Ensure that the manufacturing facilities and equipment are suitable for the intended use
- Ensure that the personnel involved in manufacturing are qualified and trained
- Ensure that the finished products are tested and meet the required standards
- Ensure that the drugs are labeled and packed properly
1. General Requirements
Location and surrounding:
- To prevent or control cross-contamination from an external source such as open sew, drains, and the public lavatory.
- From any factory that is evolving to produce obnoxious odor, fumes, dust, smoke, chemical, and biological discharge.
Building and premises:
- The building should be designed in such a way that drugs can be manufactured in hygienic conditions.
Premises:
- Premises must be compatible and have sufficient space to prevent the entry of insects, pests, and birds, interior shall be cracked-free, sufficient AHU system should be installed to recirculate fresh air. lightning LUX intensity should be as per regulation. A sufficient drainage system should be avoided from open channels.
Water system:
- It must follow as per B.I.S (beauro of Indian standard) or local municipality to produce potable/ purification water as per pharmacopeia specification.
- Purified water must be used for all manufacturing purposes and for washing.
- Must be stored in the tank to prevent any types of microbial growth.
- Cleaning must be done and records must be maintained.
Related: Water system validation
Disposal of waste:
- Waste like; waste from ETP, sewage, solids, and liquids must be disposed of as per E.P.C.B
- Biomedical water must be disposed of by biomedical waste management 1996.
- Proper management of rejected drugs and non-recoverable drugs. Records must be maintained as scheduled.
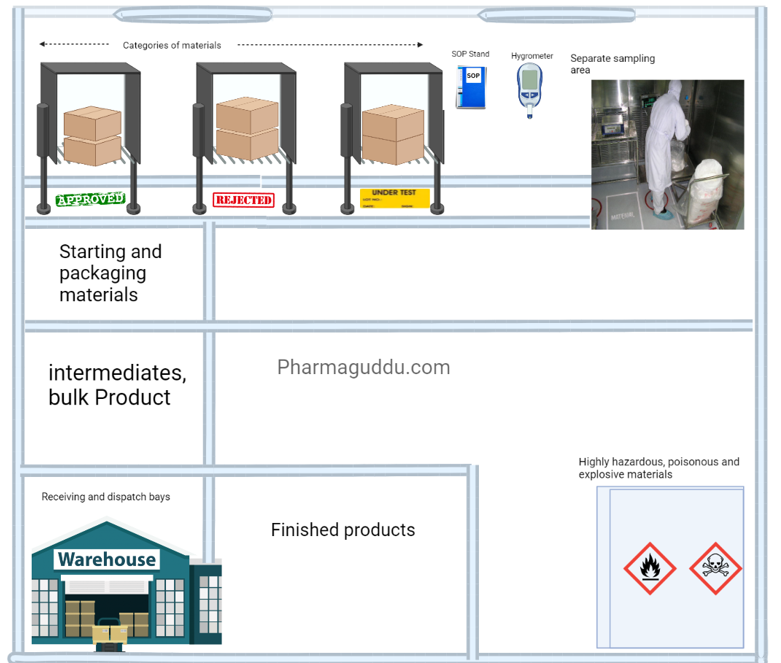
2. Warehouse area:
An abundance of spaces must be planned to allow for the sufficient and orderly warehousing of a variety of materials and goods, including raw materials, intermediates, bulk and finished goods, products that have been released from quarantine, rejected, returned, or recalled, as well as machine and equipment spare parts and replacement parts.
In short, the warehouse area temperature and humidity control shall be provided monitored, and controlled. the dispensing area must be separated, Dispatch area must be controlled by external weather, rodents, flies, and mosquitoes. There shall be a separate area for the flammable, and hazardous materials.
3. Production area:
The production area should be designed to minimize contamination and cross-contamination while keeping fewer manpower movements in process areas. To prevent dust accumulation, pipework, electrical fittings, ventilation openings, and similar service lines must be scheduled, fixed, and constructed. Service lines should ideally be distinguished by color, and the type of supply and flow direction should be marked or specified.
4. Ancillary area:
The ancillary area shall be separated from the other processing area.
5. Quality control area:
Quality Control Laboratories shall be independent of the production areas. Separate areas and AHU systems shall be provided each for physicochemical, biological, microbiological, or radio-isotope analysis. A separate instrument room with adequate area shall be provided for sensitive and sophisticated instruments employed for analysis.
6. Personnel:
The manufacture shall be conducted under the direct supervision of competent technical trained staff. Quality Assurance personnel shall be suitably qualified. Head of the Quality Control Laboratory, technical staff shall be direct involvement in testing,
7. Health, clothing, and sanitation of workers:
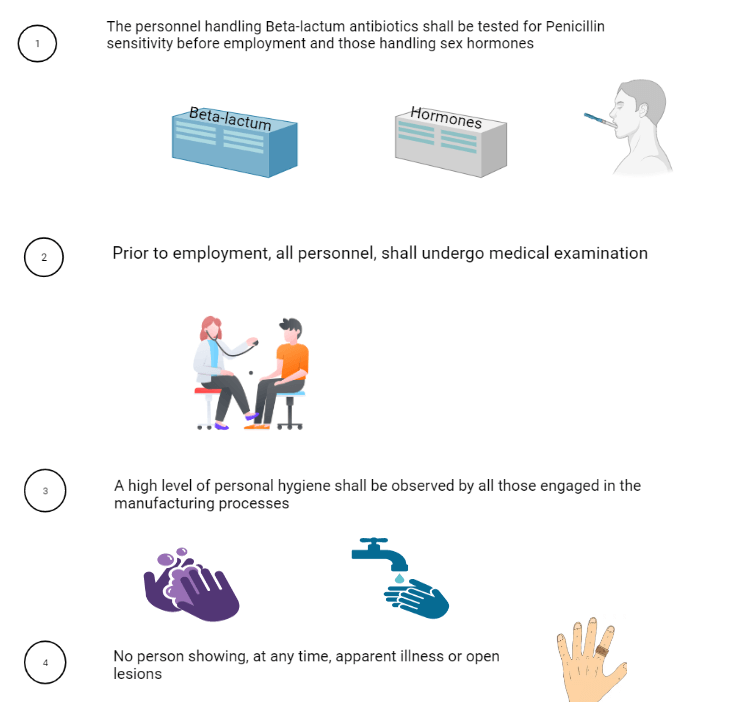
8. Manufacturing operation and control:

9. Sanitation in the manufacturing premises:
The manufacturing premises shall be:
- Cleaned
- Free from Waste and dust
- Sanitization shall be followed as per the Routine
- The procedure shall be followed
- Adequate working space to prevent mix-up and cross-contamination.
10. Raw materials
Raw Materials shall be:
- Store RM materials along with status labels on racks immediately after receipt.
- Allow only authorized persons to inter.
- Follow the FIFO rules.
- All materials shall be from approved vendors with A.R., manufacturerís name, address, and batch number; status (e.g. quarantine, under test, released, approved, rejected); the manufacturing date, expiry date, and re-test date.
- There shall be designated areas for Approved, Under test, and Rejected materials.
- Only QC-approved materials shall be used.
11. Equipment:
- The equipment shall be designed, constructed for easy operation, and cleaned while aiming to reduce the risk of cross-contamination. Defective equipment shall be removed.
- The Calibrated weighing balance shall be available in the raw material stores, production, and in process control operations, and these shall be verified and checked on a scheduled basis in accordance with SOP and records maintained.
- Equipment parts shall not be reactive, To avoid accidental contamination use non-toxic/edible-grade lubricants.
12. Documentation and Records:

13. Labels and other printed materials:
Schedule M’s requirements for labels and other printed materials aim to ensure that they are clear, accurate, and informative.

14. Quality assurance
The Quality Assurance shall ensure:
- The pharmaceutical product shall be manufactured as per GMP, GLP, and GCP.
- Control the starting, intermediate, and finished products.
- Ensure that in-process, calibration and validation are carried out as per the established procedure.
- The quality person shall be responsible for Product release and authorized to release.
15. Self-inspection and quality audit:
You may check our detailed article on “Audit Check List“
16. Quality control system:
Quality control to ensure:
- To ensure proper storage of reference samples, an adequate area with the necessary storage conditions should be provided.
- The quality control department is responsible for evaluating, maintaining, and storing these reference samples.
- SOP must be available in the area for sampling, inspecting, and testing raw materials, intermediate bulk finished products, and packing materials. Additionally, monitoring of environmental conditions may require the implementation of these procedures.
- Authorized and dated specifications for all materials, products, reagents, and solvents must be established, including tests for identity, content, purity, and quality.
- This includes specifications for water, solvents, and reagents used in the analysis. These specifications must be maintained and updated regularly to ensure compliance with Schedule M for Good Manufacturing Practice (GMP) of pharmaceuticals.
- No product shall be dispatched, or supplied until it has been certified by authorized persons.
- For each batch of products manufactured, reference or retained samples must be maintained in a quantity that is at least twice the amount of the drug required for all tests, except for sterility and pyrogen/bacterial endotoxin tests conducted on the active material and the manufactured product. The retained product must be kept in its final or simulated packaging for a period of three months after the expiration date.
- When assessing records related to finished products, all relevant factors must be considered, including production conditions, in-process testing results, manufacturing (including packaging) documentation, compliance with finished product specifications, and examination of the finished packaging. The assessment records must be signed by the production in charge and countersigned by the authorized quality control personnel before the product is released for sale or distribution. Compliance with these requirements is necessary to adhere to Schedule M for Good Manufacturing Practice (GMP).
- QC Officer must have appropriate access to production department areas for sampling and Status Labeling.
- The quality control department is responsible for conducting stability studies on products to ensure their shelf life under prescribed storage conditions. Comprehensive records of such studies must be maintained.
- The head of Quality Assurance should investigate all product complaints, and detailed records of such investigations must be kept.
- Before routine testing, all instruments must be calibrated, and testing procedures must be validated. Instruments must be periodically calibrated, and procedures must be validated.
- The Quality Control Department must approve and maintain each specification for raw materials, intermediates, final products, and packing materials. Periodic revisions of the specifications should be conducted whenever necessary.
- The Quality Control Laboratory of the licensee must have access to Pharmacopoeiae, reference standards, working standards, references, spectra, other reference materials, and technical books as required.
17. Specifications:
For raw materials and packaging materials:
- Designated name and internal code reference.
- Reference, if available, to a pharmacopoeial monograph.
- Qualitative and quantitative requirements along with acceptance limits.
- Name and address of the manufacturer or supplier, as well as the original manufacturer of the material.
- Sample of printed material.
- Instructions for sampling and testing, or references to procedures.
- Storage conditions.
- The maximum period of storage before re-testing.
Regarding product containers and closures:
- All containers and closures must adhere to pharmacopoeial requirements.
- Strictly follow suitable validated test methods, sample sizes, specifications, cleaning procedures, and sterilization procedures (where applicable) to prevent significant quality or purity issues caused by reactivity, additives, absorption, or leaching.
- Avoid using second-hand or used containers and closures.
In the case of bottles:
- Establish and adhere to a written schedule for cleaning.
- If bottles are not dried after washing, rinse them with de-ionized water or distilled water, depending on the situation.
For in-process and bulk products:
- Ensure availability of specifications for in-process material, intermediate products, and bulk products.
- Authenticate the specifications.
For finished products, appropriate specifications should include:
- Designated name and code reference of the product.
- Formula or a reference to the formula and the pharmacopoeial reference.
- Instructions for sampling and testing or references to procedures.
- Description of the dosage form and package details.
- Qualitative and quantitative requirements, including acceptance limits for release.
- Storage conditions and precautions, where applicable.
- Shelf-life.
19. Packing records:
Authorized packaging instructions must be provided for each product, pack size, and type. These instructions should include or refer to the following details:
(a) Product name.
(b) Description of the dosage form, strength, and composition.
(c) Pack size, specified in terms of the number of doses, weight, or volume of the product in the final container.
(d) Comprehensive list of all packaging materials required for a standard batch, including quantities, sizes, and types. Each packaging material should have a code or reference number corresponding to its specifications.
(e) Reproduction of relevant printed packaging materials and specimens that indicate the application of batch number and expiry date.
(f) Special precautions to be observed, including a thorough examination of the area and equipment to ensure line clearance before commencing operations.
(g) Description of the packaging process, including any significant subsidiary operations and the equipment to be used.
(h) Details of in-process controls, along with instructions for sampling and acceptance.
(i) Upon completion of the packing and labeling operation, reconciliation should be performed between the number of labeling and packaging units issued, the number of units labeled and packed, and any excess units returned or destroyed. Any significant or unusual discrepancies in the numbers should be thoroughly investigated before releasing the final batch.
20. Batch packing records:
For each batch or part batch processed, a batch packaging record must be maintained. This record should be based on the relevant sections of the packaging instructions, and the method of creating these records should be designed to prevent transcription errors.
Before initiating any packaging operation, it is essential to perform a thorough check and document that the equipment and workstations are free from previous products, unnecessary documents, or materials that are not required for the planned packaging operations. Additionally, the equipment should be clean and suitable for use.
21. Batch Processing Records:
There should be a Batch Processing Record available for each product. This record should be based on the relevant sections of the currently approved Master Formula. The preparation method outlined in the Master Formula should be designed to prevent transcription errors.
Prior to commencing any processing, a thorough check must be conducted and recorded to ensure that the equipment and workstations are free from previous products, unnecessary documents, or materials that are not required for the planned process. Additionally, it should be verified that the equipment is clean and suitable for use.
Throughout the processing, the following information should be recorded at the time each action is taken. The record should be dated and signed by the individual responsible for the processing operations:
(a) Product name
(b) Batch number being manufactured
(c) Dates and times of commencement, significant intermediate stages, and completion of production
(d) Initials of the operator involved in different significant production steps, and if applicable, the person who checked each operation
(e) Batch number and/or analytical control number, along with the quantities of each starting material actually weighed
(f) Description of relevant processing operations or events, including the major equipment used
(g) Documentation of in-process controls, including the initials of the individual(s) performing them and the obtained results
(h) Amount of product obtained after different critical stages of manufacturing (yield)
(i) Comments or explanations for significant deviations from the expected yield limits
(j) Notes on any special problems encountered, including details with signed authorization for any deviation from the Master Formula
(k) Inclusion of any recovered or reprocessed material, along with references to the recovery or reprocessing stages
22. Receipt of Materials:
Written SOPs and records should be established for the receipt of each delivery of raw materials, primary packaging materials, and printed packaging materials.
The records of the receipts should include the following information:
(a) Name of the material stated on the delivery note and the number of containers.
(b) Date of receipt.
(c) Manufacturer’s and/or supplier’s name.
(d) Manufacturer’s batch or reference number.
(e) Total quantity and number of containers, along with the quantity in each container received.
(f) Control reference number assigned after receipt.
(g) Any other relevant comments or information.
Written SOPs should be in place for the internal labeling, quarantine, and storage of starting materials, packaging materials, and other applicable materials.
SOPs should be available for each instrument and equipment, and these should be placed in close proximity to the respective instrument and equipment.
Sampling: Written SOPs for sampling should be established, including the authorization of personnel responsible for taking samples.
The sampling instructions should include the following:
(a) Method and sampling plan.
(b) Equipment to be used.
(c) Precautions to avoid contamination or deterioration of the material’s quality.
(d) Quantity of samples to be taken.
(e) Instructions for sub-division or pooling of samples if required.
(f) Types of sample containers to be used.
(g) Specific precautions, particularly for sampling sterile and hazardous materials.
Batch Numbering: SOPs should describe the details of batch (lot) numbering, ensuring that each batch of intermediate, bulk, or finished product is assigned a specific batch number.
Batch numbering SOPs applied to a processing stage and the respective packaging stage should be the same or traceable, demonstrating their connection to one homogeneous mix.
Batch number allocation should be promptly recorded in a logbook or electronic data processing system. The record should include the date of allocation, product identity, and batch size.
Testing: Procedures should be documented for testing materials and products at various stages of manufacture, including the methods and equipment to be used. The test results should be recorded.
Records of Analysis: The records should include the following data:
(a) Name of the material or product and its dosage form.
(b) Batch number and, if applicable, manufacturer and/or supplier information.
(c) Reference to relevant specifications and testing procedures.
(d) Test results, including observations, calculations, and references to any specifications or limits.
(e) Dates of testing.
(f) Initials of the individuals who performed the testing.
(g) Initials of the individuals who verified the testing and detailed calculations.
(h) Statement of release or rejection.
(i) Signature and date of the designated responsible person.
Written SOPs and associated records should be maintained for the following:
(a) Equipment assembly and validation.
(b) Analytical apparatus and calibration.
(c) Maintenance, cleaning, and sanitation.
(d) Personnel, including qualification, training, clothing, and hygiene.
(e) Environmental monitoring.
(f) Pest control.
(g) Complaints.
(h) Recalls.
(i) Returns received.
23. Reference sample:
Each lot of every active ingredient, with sufficient quality to conduct all tests (except sterility and pyrogens/Bacterial Endotoxin Test), should be retained for a period of 3 months after the expiry date of the last batch produced from that active ingredient.
Samples of finished formulations should be stored in the same containers or containers that simulate the actual packaging used for marketing the drug.
24. Reprocessing and recoveries:
Written procedures, approved by the Quality Assurance Department, should be established in cases where reprocessing is necessary. These procedures should specify the conditions and limitations for repeating chemical reactions, and the reprocessing should undergo validation.
If a product batch needs to undergo reprocessing, the procedure should be authorized and documented. An investigation into the reasons necessitating reprocessing should be conducted, and appropriate corrective measures should be implemented to prevent recurrence. The reprocessed batch should undergo stability evaluation.
In some cases, the recovery of product residue may be permitted by incorporating it into subsequent batches of the product. This can be recorded in the master production and control records.
25. Distribution records:
Before distributing or dispatching a batch of a drug, it must be ensured that the batch has undergone proper testing, approval, and release by the quality control personnel. Pre-dispatch inspections should be conducted on a random basis for each consignment to verify that only the correct goods are being dispatched. In the case of Large Volume Parenterals, if they are stocked, detailed instructions for warehousing and stocking should be established and followed after the batch is released for distribution. Periodic audits of warehousing practices at distribution centers should be conducted, and records of these audits should be maintained. Standard Operating Procedures (SOPs) should be developed for product warehousing.
Records for distribution should be maintained in a manner that allows for the traceability of finished batches of drugs to the retention level. This facilitates prompt and complete recall of a batch if and when necessary.
26. Validation and Process validation:
Validation studies are an integral part of Good Manufacturing Practices (GMP) and should be conducted according to pre-defined protocols. These studies encompass the validation of processing, testing, and cleaning procedures.
A written report summarizing the recorded results and conclusions should be prepared, documented, and maintained.
Processes and procedures should be established based on validation studies and undergo periodic revalidation to ensure their capability to achieve the intended results. Critical processes should be validated prospectively or retrospectively.
Whenever a new Master Formula or method of preparation is adopted, steps should be taken to demonstrate its suitability for routine processing. The defined process, using the specified materials and equipment, should be shown to consistently yield a product of the required quality.
Significant changes to the manufacturing process, including modifications to equipment or materials that may impact product quality and/or process reproducibility, should undergo validation.
27. Product recalls:
Read here: Product Recalls
28. Complaints and adverse reactions:
Complaints regarding product quality will be thoroughly reviewed and documented following established procedures. Designated personnel will investigate and evaluate each complaint, maintaining records of the investigation and any remedial actions taken.
Reports of serious adverse drug reactions related to product use, including accompanying comments and documents, will be promptly reported to the relevant licensing authority.
Written procedures will outline the necessary actions to be taken, including product recall, in cases of defective products.
29. Site master file:
The licensee must create a concise document known as the Site Master File. This file will provide detailed and factual information about Good Manufacturing Practices (GMP) related to the production and/or control of pharmaceutical manufacturing preparations conducted at the licensed premises. The Site Master File should include the following:
- General information about the licensed premises and its layout.
- Description of the manufacturing activities and processes performed.
- Overview of the quality management system in place.
- Documentation of personnel qualifications and training programs.
- Details of premises and equipment used for manufacturing and control.
- Procedures for handling and storage of materials and products.
- Information on quality control and testing methods employed.
- Protocols for handling deviations, investigations, and product recalls.
- Procedures for change control and validation of processes and equipment.
- Details of the site’s environmental monitoring program.
- Information on outsourcing activities, if applicable.
- Summary of relevant licenses, certifications, and inspections.
Part 1A- Specific requirements for the manufacture of parenteral and sterile products
Part 1B- Specific requirements for the manufacturing of OSD forms (For both Tablets and capsules)
References:
- Schedule M, Indian Pharmaceutical Association
- Drug and cosmetic act Rule and Schedule M

Naresh Bhakar is the Founder and Author at Pharmaguddu.com, bringing his extensive expertise in the field of pharmaceuticals to readers worldwide. He has experience in Pharma manufacturing and has worked with top Pharmaceuticals. He has rich knowledge and provides valuable insights and data through his articles and content on Pharmaguddu.com. For further inquiries or collaborations, please don’t hesitate to reach out via email at [email protected].