
GMP & Pharmaceutical Manufacturing Expert
✓ Reviewed by: Pankaj Sharma - Quality Control Specialist
Reviewed for Quality Control accuracy, laboratory practices, analytical methods, and technical relevance
📅 Last Updated: May 31, 2026
IND, or the Investigational New Drug Application, is a U.S. permission process required to move any new medicine or therapy from laboratory research to human testing, meaning clinical trials.
For example, imagine scientists have developed a new medicine. They cannot directly test it on humans. First, they must submit proof to the U.S. drug authority through an IND application, showing that the medicine appears reasonably safe, that animal and laboratory testing has already been completed, and that a proper human trial plan is ready.
This permission request itself is called the IND application.
An important point to understand is that an IND does not mean the medicine has been approved. It is only a temporary legal permission that allows the company to begin testing the drug on humans.
In simple words, it gives permission to start human clinical trials.

What Is an IND Application?
IND application is a formal request that has submit to the U.S. FDA to obtain permission to test a new drug or therapy in human clinical trials.
In simple words, when a medicine has not yet been approved for the market, it cannot be directly given to humans. Therefore, the company or researcher must obtain special authorization from the FDA, and this process is called the IND application.
Under U.S. regulations, such as 21 CFR Part 312, the IND works as an investigational exemption, meaning a temporary legal permission granted only for research purposes.
An IND application can be submitted by any sponsor, such as a pharmaceutical company, university, government agency, or an individual researcher who takes responsibility for the clinical trial.
The sponsor’s main objective is to provide enough proper information to the FDA so it can decide whether the human trial is reasonably safe or not. The application generally includes laboratory testing data, animal study results, manufacturing details, and a complete clinical trial protocol.
If the FDA determines that the safety measures are adequate, it grants permission to begin human clinical investigations.
Types of IND Applications:
When a new medicine or drug is made, then how is permission obtained from the FDA before testing it on humans? When you make a new medicine and want to test it on humans, the FDA first asks you what exactly you want to do with this drug in the end? According to this answer, there are two main categories.
Commercial IND:
The first category is Commercial IND. This means that your plan is that after the research is completed, you will use this drug in the market, sell it to people, and do business. Just like when Pfizer made the vaccine, its goal was that it should reach all patients and they should sell it. In simple words, things related to money plus public health are called Commercial IND.
Non-Commercial IND:
The second category is Research Non-Commercial IND. This means that you are doing this study only for pure research, and for now you have nothing to do with launching it in the market, earning profit, or sales. Like when a university professor is doing a study only to understand whether this compound destroys cancer cells or not. This is simple, things related only to knowledge plus research are called Non-Commercial IND. The difference between both categories is only related to the product purpose, while the safety rules are the same for both.
Now we will talk about those situations where there is a life-threatening emergency and there is no time. Sometimes in life there is no time for proper paperwork because the patient’s life is in danger. Therefore, FDA has created special fast-track pathways.
Emergency IND:
The first is Emergency IND, meaning EIND, meaning save life first. Suppose a man has a very rare infection and no regular medicine is working. The doctor has an experimental drug that might work, but man does not have time, maybe only a few hours are left. In such a situation, the process will be that the doctor will first declare an emergency and, if possible, take consent from the patient or family. But if man is unconscious, then later is also acceptable. After that, treatment can be started without waiting for written approval from the FDA. After that, within 15 working days, the application has to be sent to the FDA. The FDA gets 30 days to raise objections, and if they do not say anything, then treatment can continue. Remember, first life, then paperwork, but the paperwork must definitely be completed later.
Expanded Access:
The second is Expanded Access, meaning Compassionate Use. Suppose someone has a rare disease and its drug is still in the clinical trial stage, meaning only selected patients are able to receive it. So that person is not in the trial, but they also urgently need the same drug. In such a case, that person or their doctor can request the FDA that “I also need this investigational drug, please allow it.” This is called Expanded Access or Compassionate Use by the FDA. This is for those patients who could not become part of the trial but have a serious need for treatment. It has a separate form and a separate review process, a little detailed, but definitely possible.
Core Components of a Complete IND Package:
When a new drug is developed and it has to be tested on humans, then a complete application has to be submitted to the FDA, which is called the IND (Investigational New Drug) Package. This is not a small form, but rather a detailed technical document that covers three broad areas: Preclinical Data, Chemistry Manufacturing & Controls (CMC), and Clinical Protocols. Let’s understand these three one by one in simple language.
Preclinical Data:
First comes Preclinical Data. This section basically justifies why we now consider this drug ready to be tested on humans. It includes pharmacology (how the drug works), toxicology (whether it is harmful or not), and safety data — which mostly comes from animal studies. The purpose is to create a preliminary safety profile so that the starting dose selection for the first human trial is scientifically sound. And yes, in 2026 the FDA is now actively encouraging New Approach Methodologies (NAMs) — such as in-vitro (testing on cells in the lab), ex-vivo (testing on tissues), in-silico (computer simulations), and chemical-based tests that can replace, reduce, or refine traditional animal testing. For example, the Bovine Corneal Opacity and Permeability test is an accepted alternative for checking eye irritation. For some small molecule drugs, instead of the standard 2-year carcinogenicity study, the Weight of Evidence approach is now also considered acceptable — meaning combined data from multiple smaller studies can also work, if it is scientifically strong.
Chemistry, Manufacturing, and Controls (CMC):
The second important component is Chemistry, Manufacturing, and Controls (CMC). This section confirms that the investigational drug you are making can be consistently manufactured with quality — and that too under Good Manufacturing Practices (GMP). It covers: how the manufacturing process works, which analytical methods will be used to test the drug, what the specifications are for the drug’s identity and purity, and the stability data that proves the drug will remain safe and effective throughout the entire trial period. If any change has to be made in the manufacturing process, then it is necessary to carefully document and justify it — because the FDA wants transparency at every step. In simple words: CMC means “Our drug will have the same quality in every batch, and we can prove it.”
Clinical Protocols:
The third and final component is Clinical Protocols. This section explains how you are designing the proposed clinical trial. It includes: the study objectives (what you want to prove), dosing regimens (how much dose, how many times, and how it should be given), inclusion and exclusion criteria (which patients can be included in the trial and which cannot), and a clear safety monitoring plan (how patients will be tracked and how side effects will be handled). If these protocols are clear, logical, and scientifically sound, then the chances of getting FDA approval to start a Phase I trial increase significantly. Remember: you have to convince the FDA that your plan is safe, ethical, and will provide useful data. There is a separate review process — it is a little detailed, but definitely possible!
Administrative Documents:
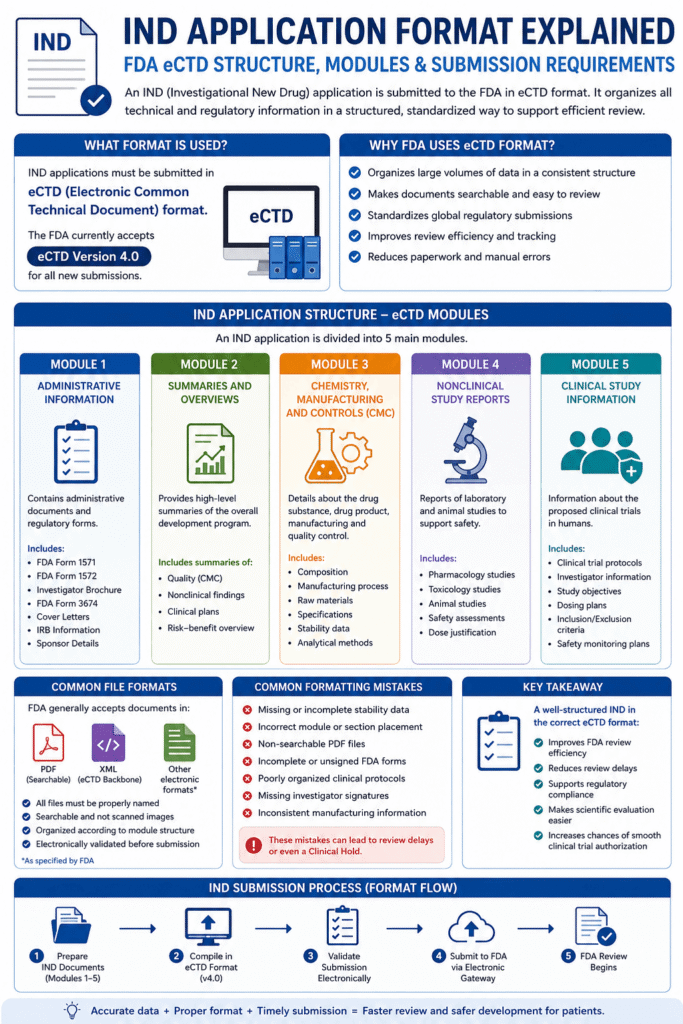
Till now, we have understood what the three main components of an IND package are — Preclinical Data, CMC, and Clinical Protocols. But only this technical content is not enough. While submitting the application to the FDA, some mandatory forms also have to be attached, and everything has to be submitted in a specific electronic format. Let’s understand all of this as well in simple language.
FDA Form-1571:
First of all, let’s talk about the forms that must be submitted along with the IND application. The first form is FDA Form-1571, which is also called the “Cover Sheet.” This is basically the front page of the application — like the title page of a school project. It contains basic information: the sponsor’s name and address (meaning the company or institution conducting the research), the drug’s name, for which disease or condition this medicine is being proposed (proposed indication), and what type of application (Commercial, Research, Emergency, etc.). In simple words, this form tells the FDA reviewer right on the first page that “What this application is about and who has sent it.”
FDA Form-3674:
The second important form is FDA Form-3674, which is called the “Investigator Brochure.” This is a compendium, meaning a complete collection — in which all the current medical and scientific information about the investigational product is collected in one place. Its main audience is investigators (the doctors or scientists who will conduct the trial) and IRBs (Institutional Review Boards, which check ethics). Think of it like giving a “Welcome Kit” to a new employee before joining, which contains all the necessary information about the company — in the same way, this brochure works as a reference guide for investigators so that they can properly understand the drug and conduct the trial effectively.
FDA Form-1572:
The third form is FDA Form-1572, meaning the “Statement of Investigator.” Every participating investigator has to personally sign this form. In this, the investigator confirms that they have the proper qualifications to conduct this trial, and that they are committed to following IND regulations. This is basically a promise letter — like when we write in an application for important work, “I will follow the rules.” For the FDA, this form is very important because it ensures that every person handling the trial is responsible and qualified.
Electronic Common Technical Document (eCTD) format:
Now let’s talk about the submission format. Earlier, applications were submitted on paper, but now everything has become digital. All IND submissions are now expected in the Electronic Common Technical Document (eCTD) format. This is a standardized digital format that ensures all documents are arranged in an organized manner, are easily searchable, and are easy for FDA reviewers to navigate.
And yes, one important update: from September 2024, the FDA started accepting eCTD version 4.0. This new version is more user-friendly, flexible, and technically advanced than before. The benefit of this is that if your submission is in this format, then the reviewer takes less time to find, compare, and review your data — because of which the overall approval process can become smooth and fast.
Related Articles around to FDA and Guidelines:
- FDA Form 483 | Warning Letters: How to Handle, Form, Example
- Pharmaceutical Quality System (PQS), ICH Q10 Guidelines
- Change Management System in Pharmaceuticals
- Complete Overview of ISO CleanRoom Classification and Risk Assessment
- Difference Between Classified and Non-Classified Areas in Pharmaceuticals
- Good Laboratory Practice (GLP) in Pharmaceutical
- What is GMP | cGMP | GMP Principle
- ICH Guidelines in Pharmaceutical (updated)
How the FDA Reviews Your IND Application and Timeline:
Till now, we have understood how an IND package is prepared, which forms are attached, and how submission is done. But now the most important question is: “What happens after the application is submitted?” Let’s understand in simple steps.
Initial 30-Day Review Period:
When your IND application gets submitted to the FDA, then a standard timeline begins. The FDA gets 30 calendar days to review it. This is a fixed waiting period — like the time taken for exam results to come out. If within these 30 days the FDA does not find any major concern in your application — neither in product quality, nor in the clinical protocol, nor in the preclinical or CMC data — then the IND automatically becomes effective after 30 days. This means that now the sponsor can ship the investigational drug to its named investigators and can formally start clinical studies. In simple words: “No news is good news” — if the FDA does not stop anything within 30 days, then you can move ahead.
Clinical Hold Decision:
But remember, the FDA can intervene at any time — whether within those 30 days or later. If the reviewers feel that there is any significant issue in the application — such as the drug quality is not proper, the clinical protocol is weak, or there are gaps in the preclinical/CMC data — then they issue a “Clinical Hold.” The simple meaning of Clinical Hold is: “Stop, first fix this problem.” If the IND has not even started yet, then this hold will delay the proposed investigation. And if the IND is already active and the trial is running, then the sponsor will have to temporarily suspend the ongoing trials. The most common reason for a clinical hold is product quality issues, after that come concerns related to clinical and nonclinical development. Once a hold is imposed, all clinical activity stops — until the sponsor fully addresses those deficiencies and the FDA formally lifts the hold.
Now let’s understand one very important concept: IND is a “Living Document.” This does not mean that once it is submitted, the work is finished. Rather, IND is a dynamic file that keeps evolving throughout the entire development journey. Sponsors have to submit amendments whenever needed — such as if any change has to be made in the protocol, a new investigator has to be added, or some new safety information has been found that needs to be reported. This process ensures that the FDA always remains updated and patient safety is never compromised.
In this context, there is a very useful tool: the Pre-IND Meeting. This is a formal opportunity where the sponsor can talk with the FDA even before submitting the application. In this meeting, you can present your non-clinical and clinical data, discuss proposed study designs, and take feedback on the overall development strategy. The biggest advantage of this is that expectations get aligned right in the beginning — because of which the risk of later deficiencies reduces, the review process becomes smooth, and the chances of getting approval increase. Think of it like showing a draft of an important project to your teacher and taking feedback before final submission — in the same way, the Pre-IND meeting is your “draft review session” with the FDA!
Clinical Trial Phases Post-IND
Till now, we have understood how an IND is submitted, how the FDA reviews it, standard timeline and how approval is granted. But now comes the most exciting part: when the IND becomes effective, clinical trials begin. Let’s understand these phases one by one.
When the IND becomes effective, clinical trials can formally begin. These trials follow a structured, phased approach — like climbing stairs: first small steps, then bigger steps. Every phase has its own purpose, and the results of each phase decide whether to move forward or not.
Phase I:
The very first step is Phase I. This is the moment when a new medicine enters the human body for the first time. The main focus of this phase is safety — meaning is this medicine safe? What dose should be given? What side effects can occur? Usually, it includes 20 to 100 participants — who can be healthy volunteers or patients as well (this depends on the therapy). In this phase, researchers also try to understand how our body processes this drug (absorption, metabolism, excretion) and how this drug affects our body (pharmacodynamics). In simple words: “First Test, First Step — Safety Comes First.”
Phase II:
If the results of Phase I are encouraging — meaning the drug appeared safe and was working with acceptable side effects — then we move forward to Phase II. In this phase, the number of participants increases — usually up to several hundred. Now the focus shifts to preliminary efficacy — meaning is this drug actually working in the target patient population for which it was made? At the same time, safety monitoring also continues. This phase is basically a “proof-of-concept.” If the results are promising, then the program moves forward. If not — if the drug does not appear effective or unexpected safety issues arise — then development can stop here itself. Simple analogy: “Phase II = Reality Check — Will It Really Work?”
Phase III:
When Phase II becomes successful, then comes the biggest and most critical step: Phase III. These are large-scale pivotal studies in which several hundred to several thousand participants are included. These trials are usually randomized and controlled — meaning some patients receive the new drug, while some receive a placebo or the existing standard treatment.
The purpose of this is to generate strong, statistically significant evidence that this drug is safe and effective. Successful Phase III data is the core evidence that is submitted to the FDA for marketing approval (NDA or BLA). And yes, one important update: the FDA now generally considers a single pivotal trial as the default for approval — therefore, the trial design and statistical rigor must be precise and strong right from the beginning. In simple words: “Phase III = Final Exam — Pass It and You Get the Degree.”
Phase IV:
And when the drug finally gets approved and enters the market, even then the journey does not end. Then comes Phase IV — which is also called post-marketing surveillance. In this phase, long-term safety and efficacy data are collected — in a larger and more diverse population, which was not possible in pre-approval trials. These studies can identify rare or delayed adverse effects, and they also help define where this drug fits in therapy — for which patients it is best, in which combinations it works, etc. Simple analogy: “Phase IV = Life After Launch — How Is It Performing in the Real World?”
Safety Reporting Obligations:
Safety reporting is not a formality. It is a core patient protection requirement that runs throughout the entire IND lifecycle.
Sponsor Responsibilities:
Till now, we have understood the phases of clinical trials. But one very important question is: “The trial is running, patients are taking the drug — if something goes wrong, then what will happen?” Let’s understand this concepts.
When a clinical trial is running, then the biggest responsibility of the sponsor (meaning the company or institution conducting the research) is to monitor the safety of the participants. Every reaction of every patient, every side effect, every small or big change — everything has to be carefully tracked. And only tracking is not enough; if something serious happens, then it also has to be reported to the FDA on time. This is not an optional task, but a strict regulatory requirement.
Serious Adverse Event (SAE):
Now here comes an important concept: Serious Adverse Event (SAE). An SAE is something that puts the patient’s life in danger, requires hospitalization, causes permanent disability, or creates any other major health issue. But not every SAE is automatically reportable. According to FDA rules, if an adverse event is serious, unexpected, and may have been caused by the investigational drug — only then is it mandatory to report it to the FDA within 15 calendar days.
unexpected adverse event:
Now the question is: “What does unexpected mean?” In simple words, an unexpected adverse event is one whose nature, severity, or outcome does not match the information already written in the Investigator Brochure. Suppose the brochure says that the drug may only cause a mild headache, but during the trial a patient develops severe liver damage — then this will be considered “unexpected.” If this event is also serious and also appears related to the drug, then the sponsor will have to formally report it to the FDA within 15 days of becoming aware of it. This timeline is strict — in calendar days, not working days. Meaning weekends and holidays will also be counted.
Investigator Responsibilities:
Till now, we have understood how the sponsor monitors safety and sends reports to the FDA. But in a clinical trial, not only the sponsor is responsible — Investigators (the doctors or scientists who actually conduct the trial) also have very big responsibilities.
First of all, let’s talk about the responsibilities of investigators. When an investigator is conducting a clinical trial, then their most important duty is to track the safety of the participants. If during the trial any unexpected serious adverse event occurs — meaning any serious health issue that was not previously expected — then it is mandatory for the investigator to report it to two places: first, the IND sponsor, and second, the Institutional Review Board (IRB). And the time limit for sending this report is strict — within a maximum of 10 working days after first becoming aware of it. This timeline is slightly different from the sponsor’s 15-calendar-day rule, therefore investigators have to keep both deadlines in mind. In simple words: “If anything serious and unexpected happens — inform two places within 10 days.”
Another important rule is that whenever any IND safety report is submitted, it must also mention whether any similar suspected adverse reaction had been reported earlier. This is called cumulative reporting — meaning every new report combines with previous reports to create a complete safety picture. This process helps build a comprehensive safety profile of the drug over time. Think of it like building a puzzle — every new piece combines with the previous pieces to make the complete image clearer. In the same way, every safety report makes the safety story of the drug clearer.
The E2B(R3) Transition:
Now let’s talk about one major technical update that became applicable from 2026: the E2B(R3) Transition. Earlier, safety reports used to be submitted in the E2B(R2) format, but now all pre-market IND safety reports must be submitted only in the ICH E2B(R3) electronic format. This new format is a globally harmonized system — meaning regulatory authorities around the world can now exchange safety data in the same standard, making communication faster and more accurate.
Full compliance became mandatory from April 1, 2026. But for those companies that had not yet upgraded their systems, there was a grace period — they could submit in the old E2B(R2) format until September 30, 2026. But remember: this grace period was only for companies already using the system — new adopters (meaning those who are newly entering this process now) will have to directly use E2B(R3). Simple analogy: “Old users got some time to update their system, but new users will have to directly use the new version.”
Independent Oversight:
In some clinical trials, there is a separate body called the Data and Safety Monitoring Board (DSMB) or Data Monitoring Committee (DMC). This board is independent from the trial team — meaning they are neutral experts separate from the people conducting the trial. DSMB is not mandatory for every trial, but the FDA strongly recommends it for trials where:
- Adaptive designs are being used (meaning changes can be made during the trial),
- Trials are being conducted on pediatric populations (children),
- Or the participant risk level is elevated.
The job of the DSMB is to regularly review the accumulating data and, based on that data, recommend whether the trial should continue, should be modified, or if safety concerns are very serious, should even be terminated. This acts as an extra layer of protection for participants — like calling an external auditor for an important project so that there is no bias.
Common Challenges in IND and How to Overcome Them
One important question is: “If preparing an IND is so important, then where do people make mistakes?” Today let’s talk about those common failure patterns that repeatedly occur — and most importantly, how they can be avoided.
Incomplete or Inconsistent Data Packages:
The first and most common mistake is: Incomplete or Inconsistent Data Packages. Imagine you want to build a house, but the foundation plans are incomplete, the quality of the bricks is not clear, and the architect’s design is also somewhat confusing — then will the builder start the work? Absolutely not. In the same way, when there are gaps in the CMC (Chemistry, Manufacturing, Controls) or preclinical sections of the IND application, then the FDA starts having concerns.
For example: the drug purity data is incomplete, the stability testing results are missing, consistency is not visible in the manufacturing process, or it is not clear that “Why did you choose this starting dose for humans?” The combined effect of all these small things is a clinical hold — meaning the trial gets stopped even before it starts. Simple tip: “Treat every section like a puzzle — if one piece is missing, then the complete picture will not be clear.”
Poorly Structured Clinical Protocols:
The second common mistake is: Poorly Structured Clinical Protocols. Suppose you planned a trip, but the destination is not clear, the route map is incomplete, and no emergency plan has been made — then will you be able to confidently go on the trip? No, right? In the same way, if the clinical trial protocol is ambiguous — the study objectives are not clear, the safety monitoring plan is weak, or the inclusion/exclusion criteria are confusing — then the FDA is not convinced that this trial is scientifically sound and ethically safe. The result? Either approval gets delayed, or you are asked to revise the protocol, in which both time and money get wasted. Simple tip: “While writing the protocol, ask yourself: Can a new investigator read this and conduct the trial without confusion?”
Complex Therapies:
The third and increasingly important challenge is: Complex Therapies that require Specialized Knowledge. Nowadays it is the era of cell therapies, gene therapies, and advanced biologics — and these are completely different from traditional small-molecule drugs.
In these, safety considerations are new (such as immune response, long-term genetic effects), manufacturing is complex (such as handling living cells), and regulatory expectations are also evolving. Therefore, if you are working on such a complex therapy, then it is necessary to include Intellectual Property (IP) and Regulatory teams in the Phase I design discussions right from the beginning — not after submission. Because if you do not clarify IP protection or regulatory strategy in the beginning itself, then making changes later can become very costly and time-consuming.
Modern Strategies That Strengthen IND Submissions:
When it comes to developing new medicines, depending only on traditional methods has now become outdated. Nowadays, rapidly evolving technology and smart strategies are being used in drug development, which not only save time, but also make research more accurate, cost-effective, and patient-friendly.
Model-Informed Drug Development (MIDD):
The first concept is Model-Informed Drug Development (MIDD). Its simple meaning is that nowadays researchers use mathematical and statistical models to combine preclinical (lab/animal) and clinical (human) data together. Earlier, the approach was to find the “maximum tolerated dose” — meaning the dose that is safest for the patient at the highest level.
But now, with the help of MIDD, we focus on the “optimal biological dose” — meaning the dose that actually provides therapeutic benefit without unnecessary side effects. These models scientifically optimize both dose selection and patient selection (patient enrichment), which greatly reduces the risk of expensive late-phase trials. Think of it like using GPS to choose the best route according to traffic conditions; in the same way, MIDD predicts the best development path through data patterns.
Adaptive Trial Designs:
The second important update is Adaptive Trial Designs. Traditional clinical trials used to follow a fixed plan — at the beginning itself, it was decided how many patients would be included, what dose would be given, and that same rigid plan would be followed until the end. But in adaptive design, flexibility is already embedded into the protocol — meaning if the accumulating data during the trial shows a different trend, then some things can legally and ethically be changed. For example, increasing or decreasing the sample size, adjusting the dosing regimen, or stopping weak arms early. The best part is that these changes are pre-planned and do not compromise statistical validity.
When implemented correctly, adaptive trials significantly shorten the overall development timeline. And yes, the ICH E20 guideline being developed for adaptive trials will soon be finalized, due to which its use will become a harmonized international standard across the world. In simple words: “Do not make the trial rigid, intelligently adjust according to the data.”
New Approach Methodologies (NAMs):
The third and very impactful shift is the use of New Approach Methodologies (NAMs). Earlier, every new drug had to heavily depend on animal testing, but now the use of in-vitro assays (testing on human cells/tissues in the lab), computational modeling (advanced computer simulations), and other alternatives has increased rapidly. These methods are faster, more ethical, and more cost-effective compared to traditional animal testing, and they generate high-quality safety data.
The FDA is now officially supporting this shift — for example, instead of lengthy 2-year carcinogenicity studies, the “Weight of Evidence” approach is now being accepted, where combined data from multiple smaller but scientifically strong studies is sufficient. Another excellent example is the CiPA initiative, in which hiPSC (human induced pluripotent stem cells)-based cardiac safety assessments are being included in IND packages. This means that now, instead of animal hearts, we can accurately predict drug-induced heart risks through human-derived cells. This is not just a technology upgrade, but an ethical and scientific revolution that is making drug development faster and more humane.
The common goal of all these three approaches is the same: to make drug development smarter, faster, and safer. MIDD makes data intelligent, adaptive trials make trials flexible, and NAMs make testing modern and scientifically robust. When you combine them correctly, not only does the FDA review process become smooth, but life-saving treatments also reach patients faster.
IND Application Format Explained: FDA eCTD Structure, Modules & Submission Requirements:
Conclusion:
The IND process is not just paperwork, but a structured journey that creates a balance between patient safety and scientific innovation. Proper planning, complete data, clear protocols, and timely reporting — all of these together make the path to FDA approval smooth. Nowadays, modern tools like MIDD, adaptive trials, and NAMs are making this journey smarter, faster, and more ethical. Remember: “Following the rules is important, but understanding the ‘why’ behind them is even more important.” When you prioritize patient safety and keep science evidence-based, then approval automatically comes closer.

Naresh Bhakar is the Founder and Author at Pharmaguddu.com, bringing his extensive expertise in the field of pharmaceuticals to readers worldwide. He has experience in Pharma manufacturing and has worked with top Pharmaceuticals. He has rich knowledge and provides valuable insights and data through his articles and content on Pharmaguddu.com. For further inquiries or collaborations, please don’t hesitate to reach out via email at [email protected].